

Establishing Computational Approaches Towards Identifying Malarial Allosteric Modulators: A Case Study of Plasmodium falciparum Hsp70s
- PMID: 31717270
- PMCID: PMC6887781
- DOI: 10.3390/ijms20225574
Establishing Computational Approaches Towards Identifying Malarial Allosteric Modulators: A Case Study of Plasmodium falciparum Hsp70s
Abstract
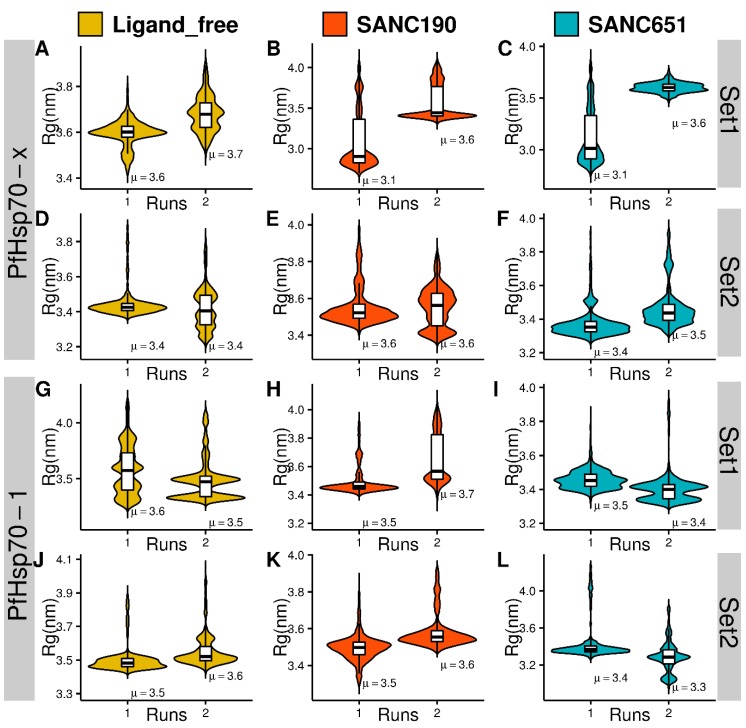
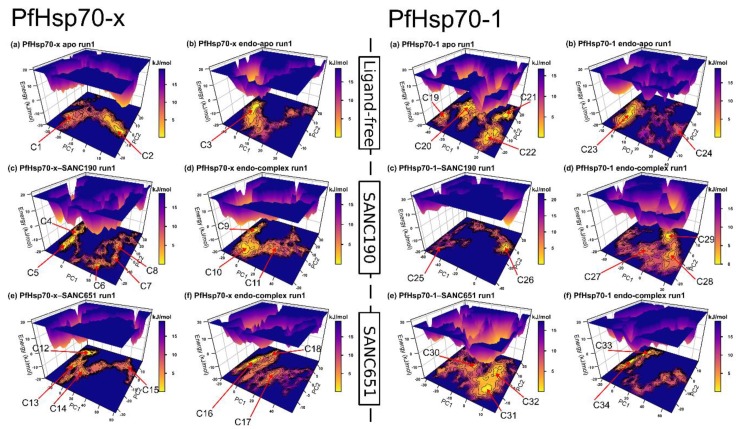
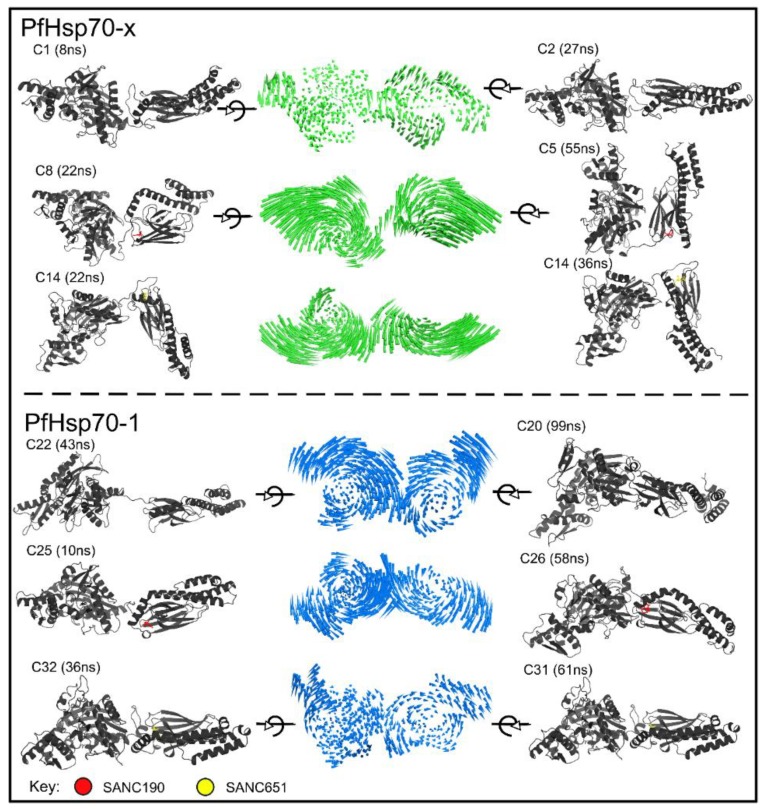
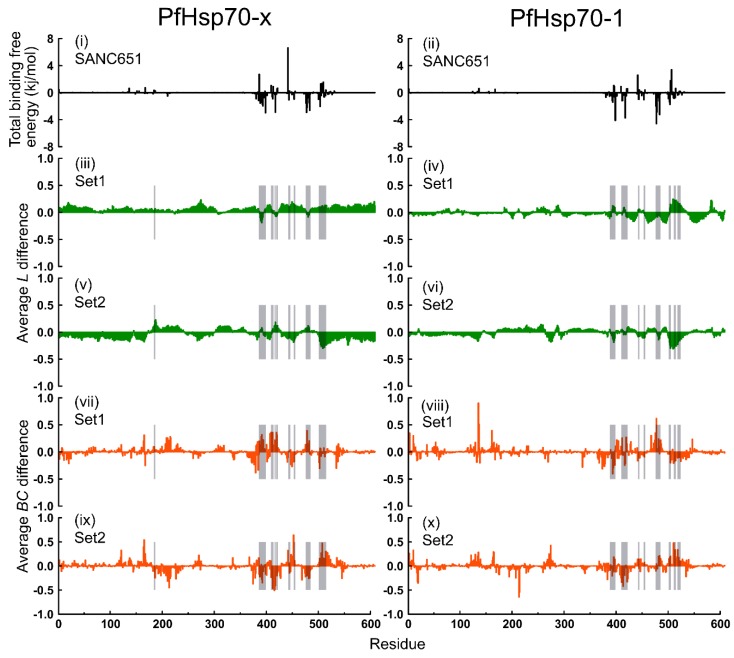
Combating malaria is almost a never-ending battle, as Plasmodium parasites develop resistance to the drugs used against them, as observed recently in artemisinin-based combination therapies. The main concern now is if the resistant parasite strains spread from Southeast Asia to Africa, the continent hosting most malaria cases. To prevent catastrophic results, we need to find non-conventional approaches. Allosteric drug targeting sites and modulators might be a new hope for malarial treatments. Heat shock proteins (HSPs) are potential malarial drug targets and have complex allosteric control mechanisms. Yet, studies on designing allosteric modulators against them are limited. Here, we identified allosteric modulators (SANC190 and SANC651) against P. falciparum Hsp70-1 and Hsp70-x, affecting the conformational dynamics of the proteins, delicately balanced by the endogenous ligands. Previously, we established a pipeline to identify allosteric sites and modulators. This study also further investigated alternative approaches to speed up the process by comparing all atom molecular dynamics simulations and dynamic residue network analysis with the coarse-grained (CG) versions of the calculations. Betweenness centrality (BC) profiles for PfHsp70-1 and PfHsp70-x derived from CG simulations not only revealed similar trends but also pointed to the same functional regions and specific residues corresponding to BC profile peaks.
Keywords: South African natural compounds; allosteric drugs; betweenness centrality; dynamic residue networks; heat shock proteins.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationship that could be construed as a potential conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
Figures
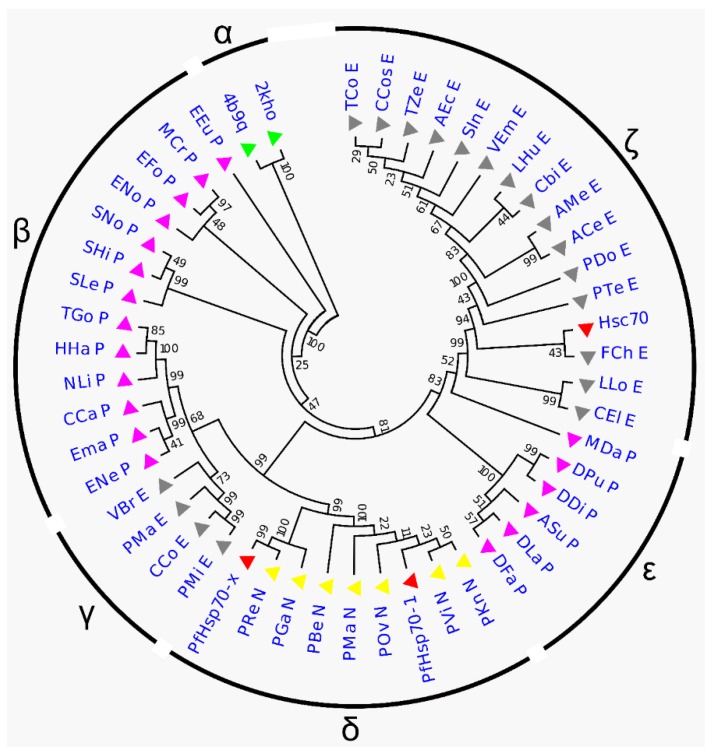
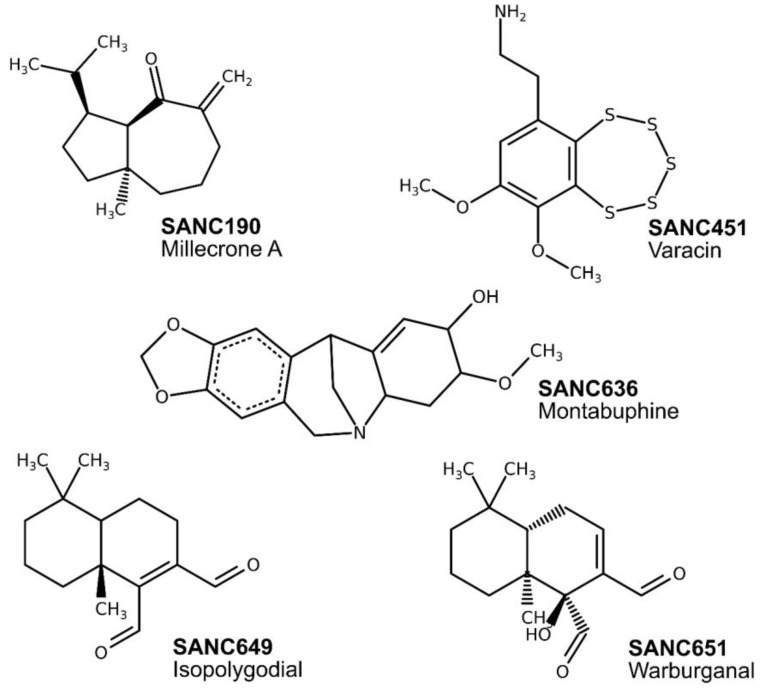
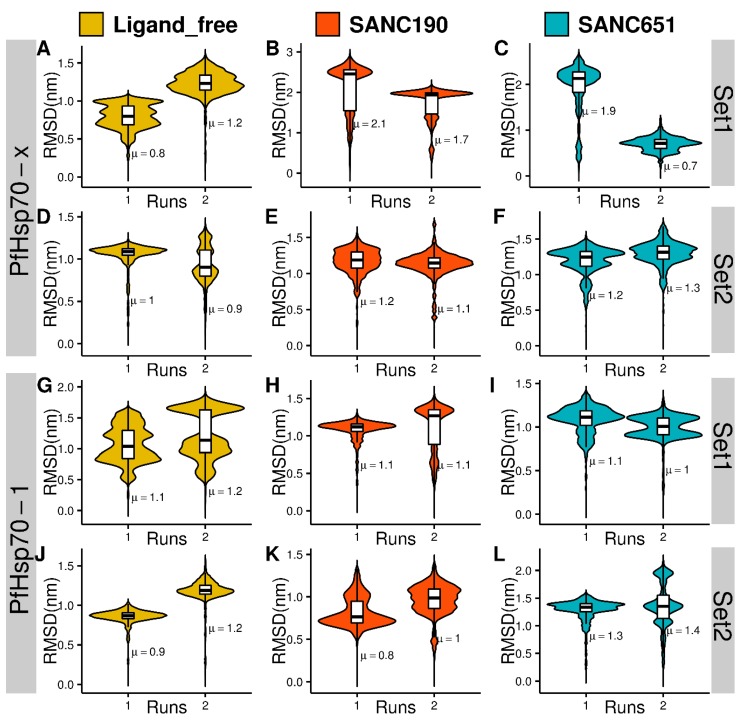
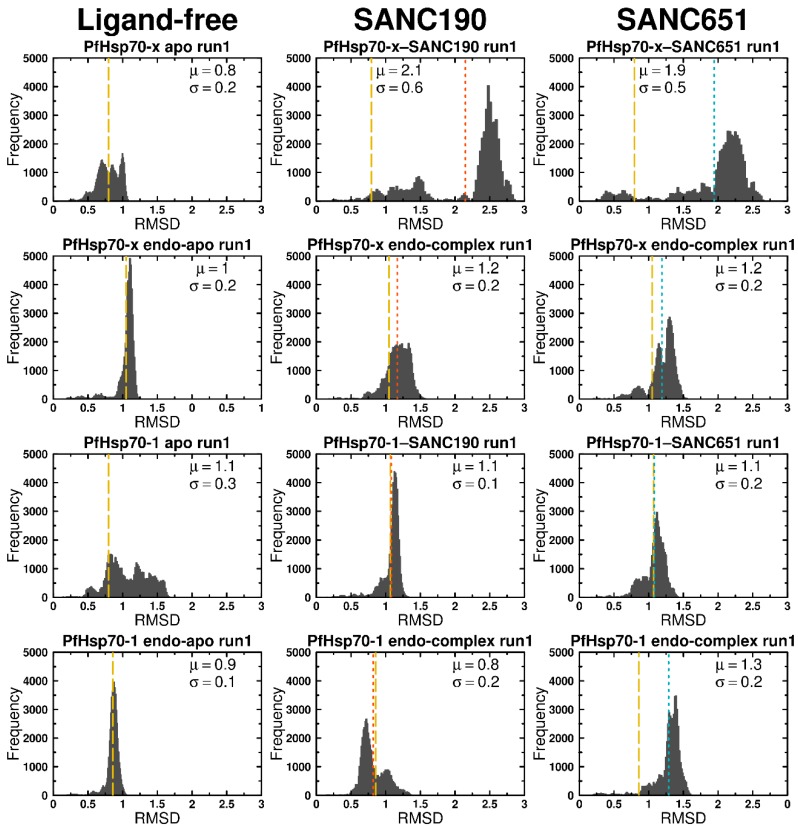

References
-
- Organization W.H. World Malaria Report 2018. World Health Organization; Geneva, Switzerland: 2018.
-
- Musyoka T.M., Kanzi A.M., Lobb K.A., Tastan Bishop Ö. Analysis of non-peptidic compounds as potential malarial inhibitors against Plasmodial cysteine proteases via integrated virtual screening workflow. J. Biomol. Struct. Dyn. 2016;34:2084–2101. doi: 10.1080/07391102.2015.1108231. - DOI - PMC - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
