

In Silico Study of Rett Syndrome Treatment-Related Genes, MECP2, CDKL5, and FOXG1, by Evolutionary Classification and Disordered Region Assessment
- PMID: 31717404
- PMCID: PMC6888432
- DOI: 10.3390/ijms20225593
In Silico Study of Rett Syndrome Treatment-Related Genes, MECP2, CDKL5, and FOXG1, by Evolutionary Classification and Disordered Region Assessment
Abstract
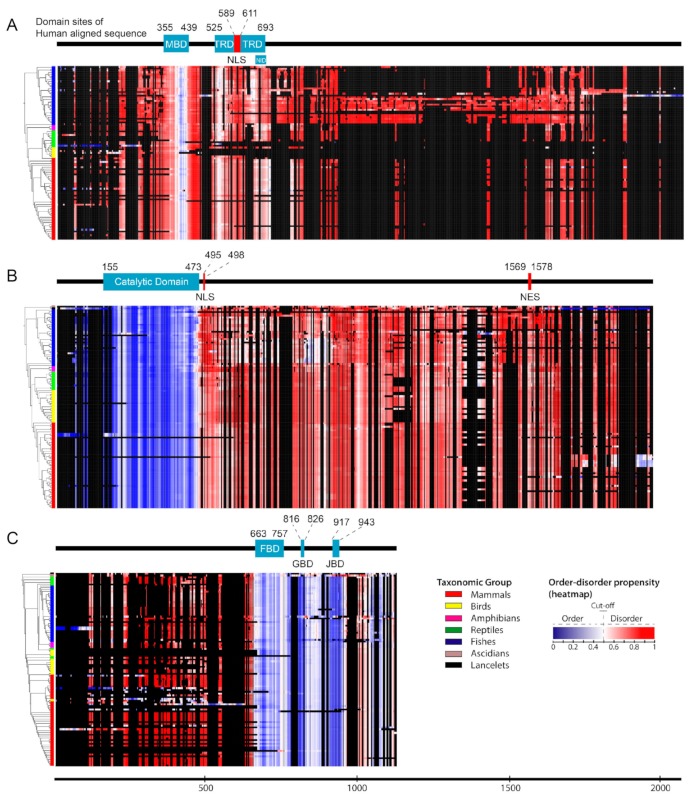
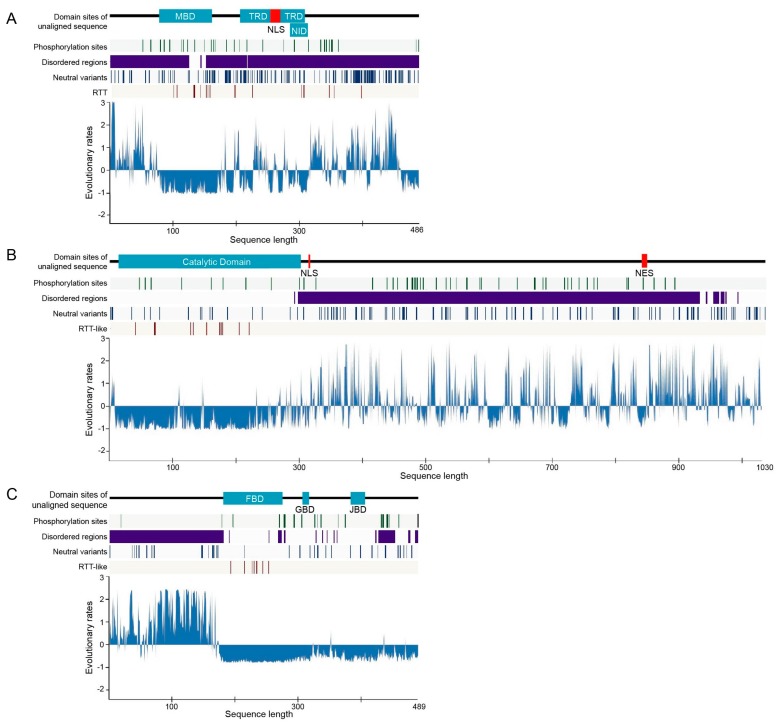
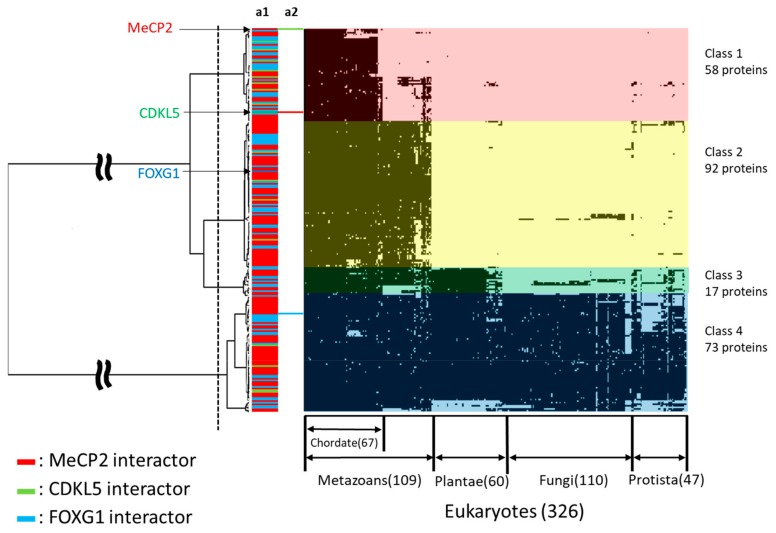
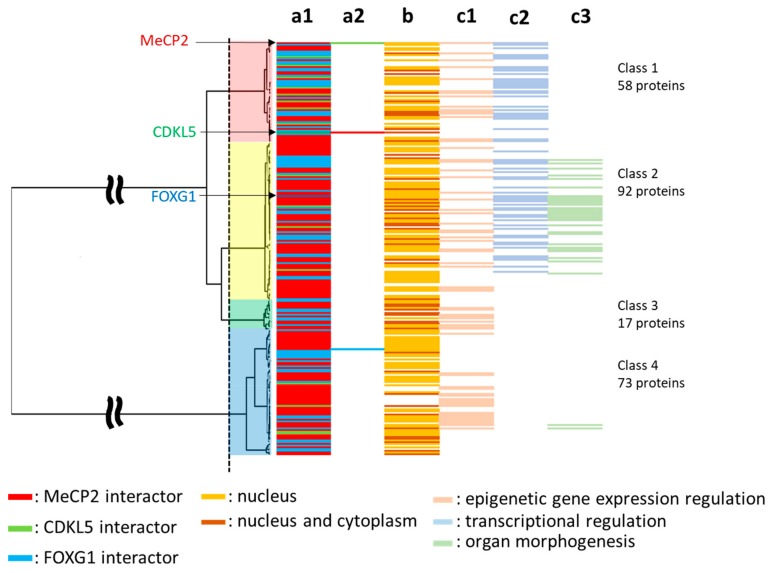
Rett syndrome (RTT), a neurodevelopmental disorder, is mainly caused by mutations in methyl CpG-binding protein 2 (MECP2), which has multiple functions such as binding to methylated DNA or interacting with a transcriptional co-repressor complex. It has been established that alterations in cyclin-dependent kinase-like 5 (CDKL5) or forkhead box protein G1 (FOXG1) correspond to distinct neurodevelopmental disorders, given that a series of studies have indicated that RTT is also caused by alterations in either one of these genes. We investigated the evolution and molecular features of MeCP2, CDKL5, and FOXG1 and their binding partners using phylogenetic profiling to gain a better understanding of their similarities. We also predicted the structural order-disorder propensity and assessed the evolutionary rates per site of MeCP2, CDKL5, and FOXG1 to investigate the relationships between disordered structure and other related properties with RTT. Here, we provide insight to the structural characteristics, evolution and interaction landscapes of those three proteins. We also uncovered the disordered structure properties and evolution of those proteins which may provide valuable information for the development of therapeutic strategies of RTT.
Keywords: Rett syndrome; cyclin-dependent kinase-like 5; forkhead box protein G1; intrinsically disordered region; methyl-CpG-binding protein 2; phylogenetic profile analysis; post-transcriptional modification.
Conflict of interest statement
The authors declare no competing interest.
Figures
References
-
- Rett A. On a unusual brain atrophy syndrome in hyperammonemia in childhood. Wien. Med. Wochenschr. 1966;116:723–726. - PubMed
-
- Weaving L.S., Christodoulou J., Williamson S.L., Friend K.L., McKenzie O.L., Archer H., Evans J., Clarke A., Pelka G.J., Tam P.P., et al. Mutations of CDKL5 cause a severe neurodevelopmental disorder with infantile spasms and mental retardation. Am. J. Hum. Genet. 2004;75:1079–1093. doi: 10.1086/426462. - DOI - PMC - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
