

Perturbation of Methionine/S-adenosylmethionine Metabolism as a Novel Vulnerability in MLL Rearranged Leukemia
- PMID: 31717699
- PMCID: PMC6912509
- DOI: 10.3390/cells8111322
Perturbation of Methionine/S-adenosylmethionine Metabolism as a Novel Vulnerability in MLL Rearranged Leukemia
Abstract
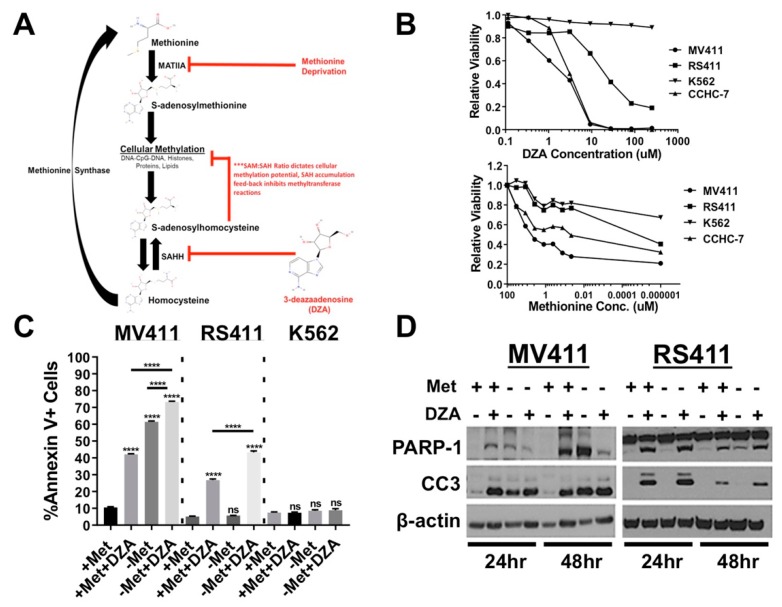
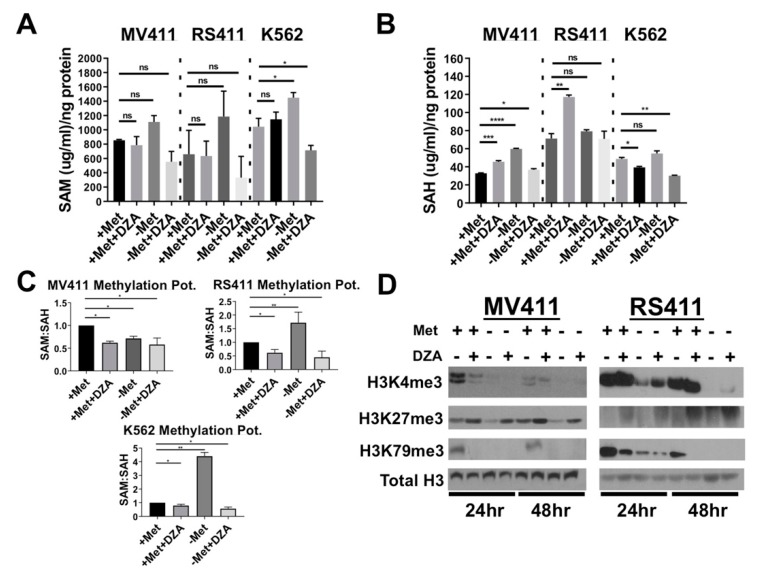
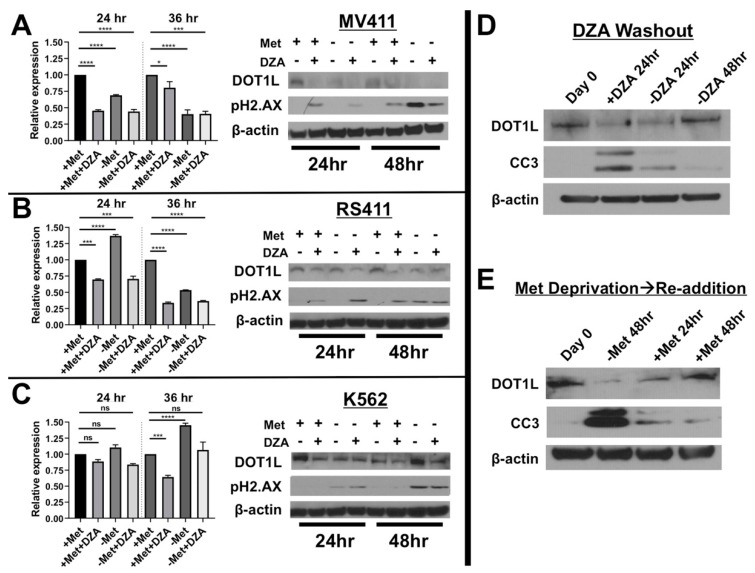
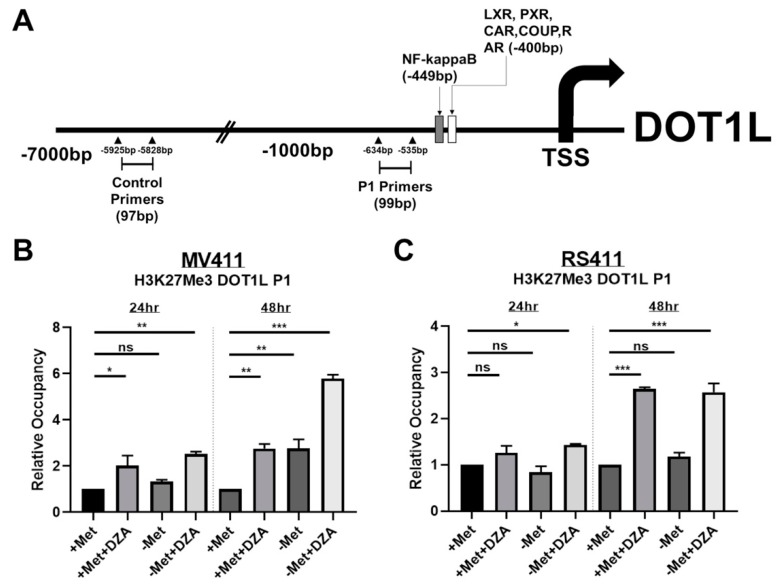
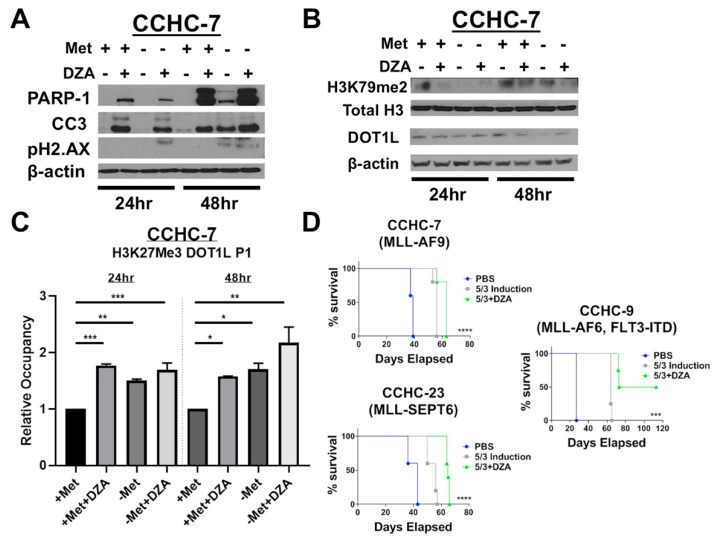
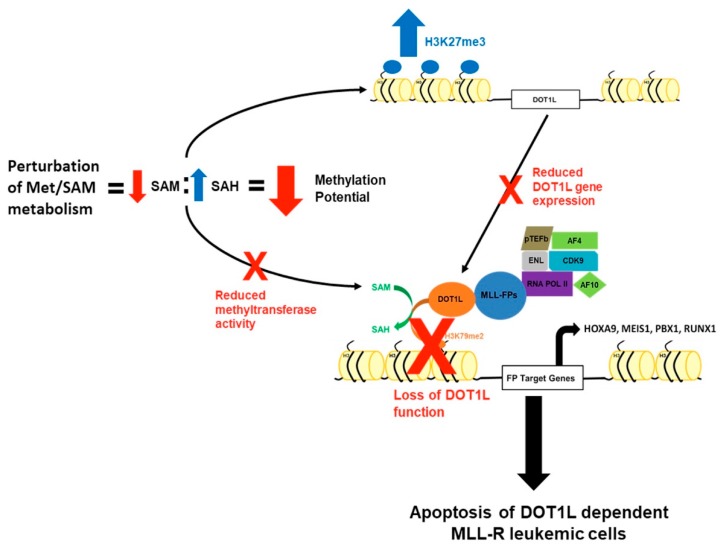
Leukemias bearing mixed lineage leukemia (MLL) rearrangement (MLL-R) resulting in expression of oncogenic MLL fusion proteins (MLL-FPs) represent an especially aggressive disease subtype with the worst overall prognoses and chemotherapeutic response. MLL-R leukemias are uniquely dependent on the epigenetic function of the H3K79 methyltransferase DOT1L, which is misdirected by MLL-FPs activating gene expression, driving transformation and leukemogenesis. Given the functional necessity of these leukemias to maintain adequate methylation potential allowing aberrant activating histone methylation to proceed, driving leukemic gene expression, we investigated perturbation of methionine (Met)/S-adenosylmethionine (SAM) metabolism as a novel therapeutic paradigm for MLL-R leukemia. Disruption of Met/SAM metabolism, by either methionine deprivation or pharmacologic inhibition of downstream metabolism, reduced overall cellular methylation potential, reduced relative cell numbers, and induced apoptosis selectively in established MLL-AF4 cell lines or MLL-AF6-expressing patient blasts but not in BCR-ABL-driven K562 cells. Global histone methylation dynamics were altered, with a profound loss of requisite H3K79 methylation, indicating inhibition of DOT1L function. Relative occupancy of the repressive H3K27me3 modification was increased at the DOT1L promoter in MLL-R cells, and DOT1L mRNA and protein expression was reduced. Finally, pharmacologic inhibition of Met/SAM metabolism significantly prolonged survival in an advanced, clinically relevant patient-derived MLL-R leukemia xenograft model, in combination with cytotoxic induction chemotherapy. Our findings provide support for further investigation into the development of highly specific allosteric inhibitors of enzymatic mediators of Met/SAM metabolism or dietary manipulation of methionine levels. Such inhibitors may lead to enhanced treatment outcomes for MLL-R leukemia, along with cytotoxic chemotherapy or DOT1L inhibitors.
Keywords: AML; MLL; Methionine; SAM; mouse model.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
