Divergence dating using mixed effects clock modelling: An application to HIV-1
- PMID: 31720009
- PMCID: PMC6830409
- DOI: 10.1093/ve/vez036
Divergence dating using mixed effects clock modelling: An application to HIV-1
Abstract
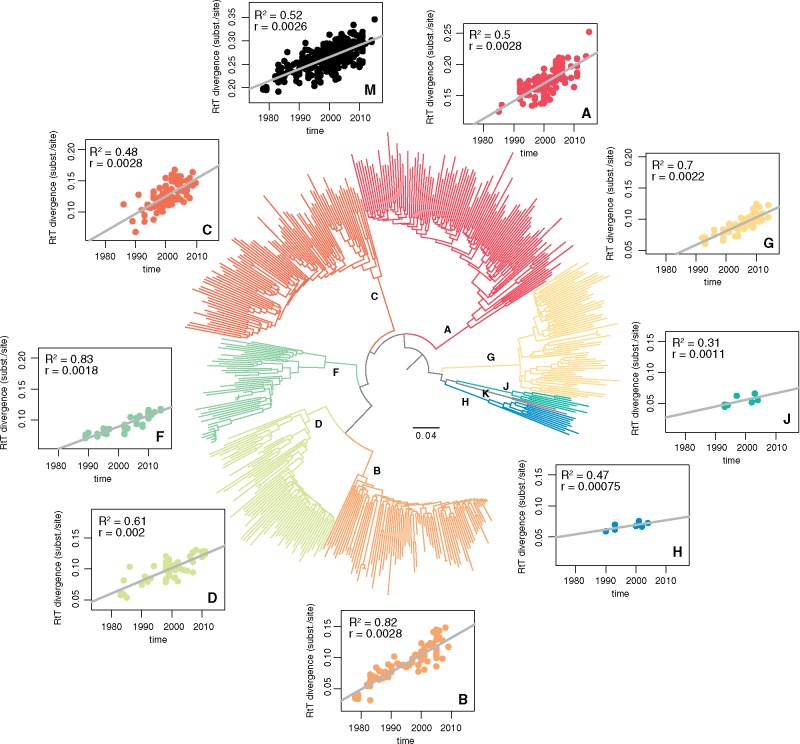
The need to estimate divergence times in evolutionary histories in the presence of various sources of substitution rate variation has stimulated a rich development of relaxed molecular clock models. Viral evolutionary studies frequently adopt an uncorrelated clock model as a generic relaxed molecular clock process, but this may impose considerable estimation bias if discrete rate variation exists among clades or lineages. For HIV-1 group M, rate variation among subtypes has been shown to result in inconsistencies in time to the most recent common ancestor estimation. Although this calls into question the adequacy of available molecular dating methods, no solution to this problem has been offered so far. Here, we investigate the use of mixed effects molecular clock models, which combine both fixed and random effects in the evolutionary rate, to estimate divergence times. Using simulation, we demonstrate that this model outperforms existing molecular clock models in a Bayesian framework for estimating time-measured phylogenies in the presence of mixed sources of rate variation, while also maintaining good performance in simpler scenarios. By analysing a comprehensive HIV-1 group M complete genome data set we confirm considerable rate variation among subtypes that is not adequately modelled by uncorrelated relaxed clock models. The mixed effects clock model can accommodate this rate variation and results in a time to the most recent common ancestor of HIV-1 group M of 1920 (1915-25), which is only slightly earlier than the uncorrelated relaxed clock estimate for the same data set. The use of complete genome data appears to have a more profound impact than the molecular clock model because it reduces the credible intervals by 50 per cent relative to similar estimates based on short envelope gene sequences.
Keywords: Bayesian inference; HIV; divergence time; mixed effects; molecular clock.
© The Author(s) 2019. Published by Oxford University Press.
Figures