

The effects of soil phosphorus content on plant microbiota are driven by the plant phosphate starvation response
- PMID: 31721759
- PMCID: PMC6876890
- DOI: 10.1371/journal.pbio.3000534
The effects of soil phosphorus content on plant microbiota are driven by the plant phosphate starvation response
Abstract
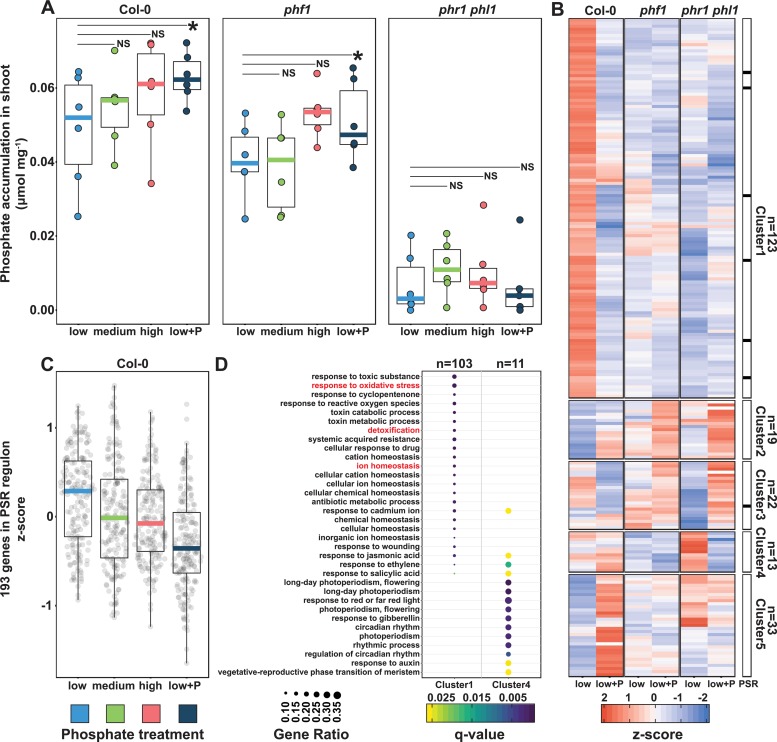
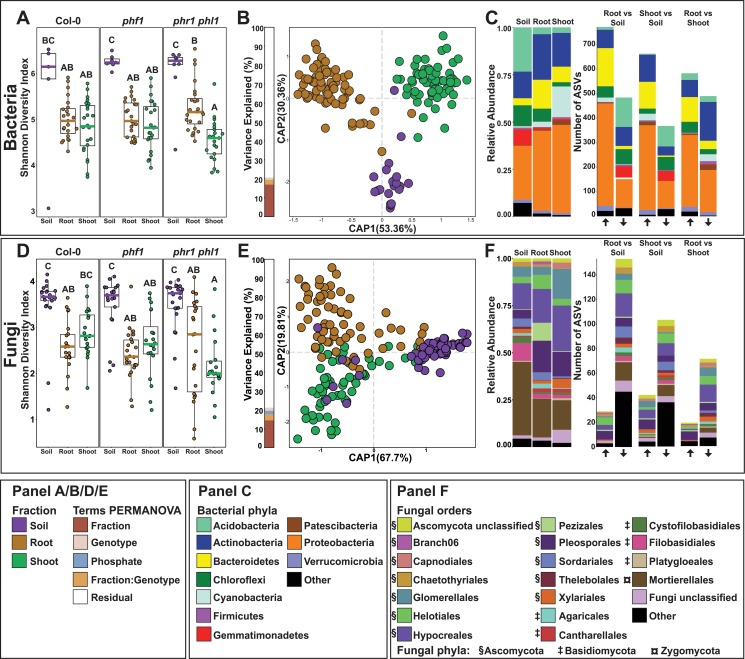
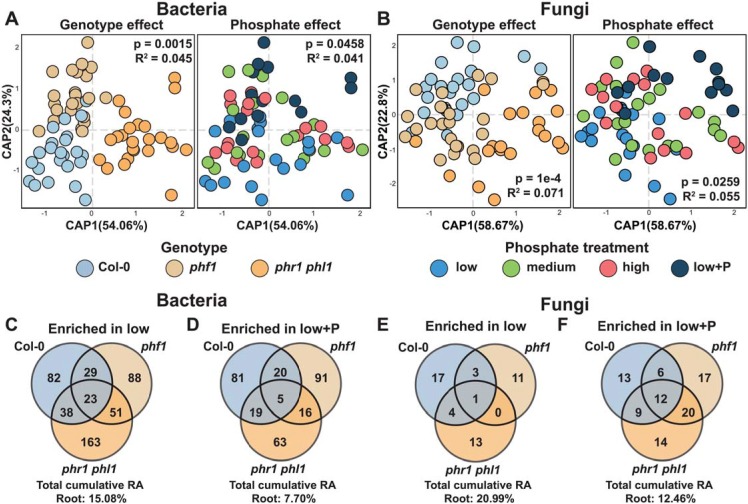
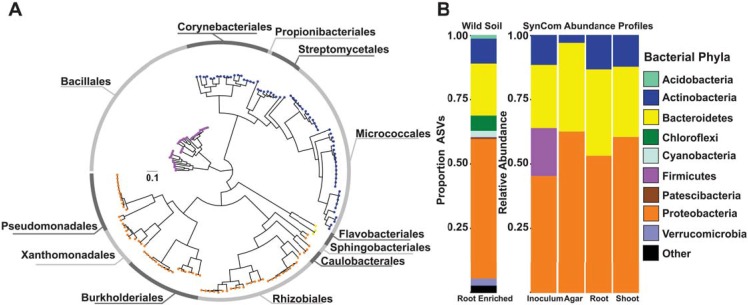
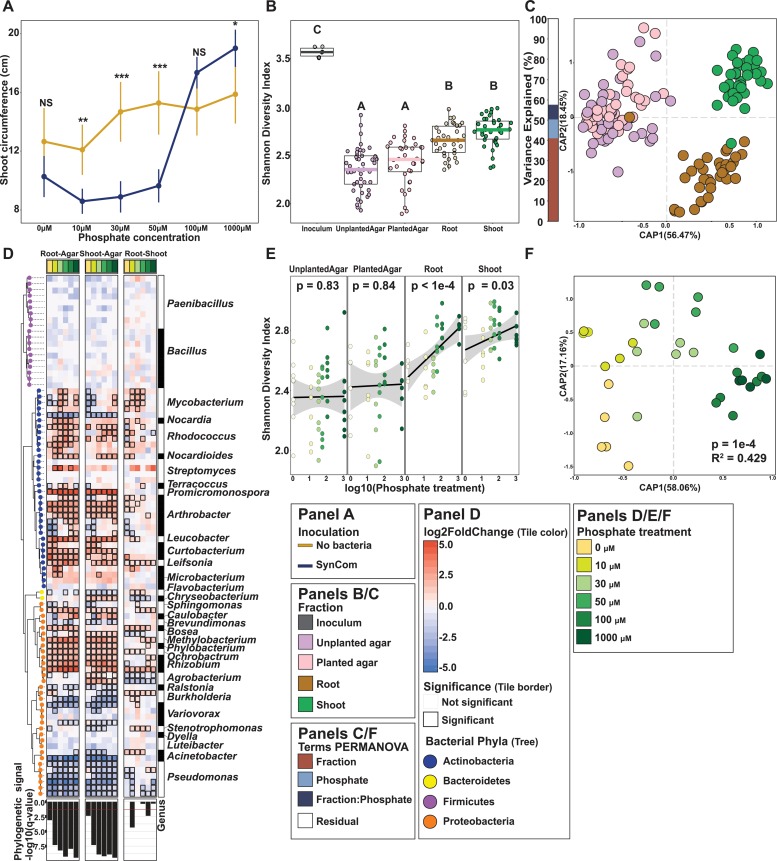
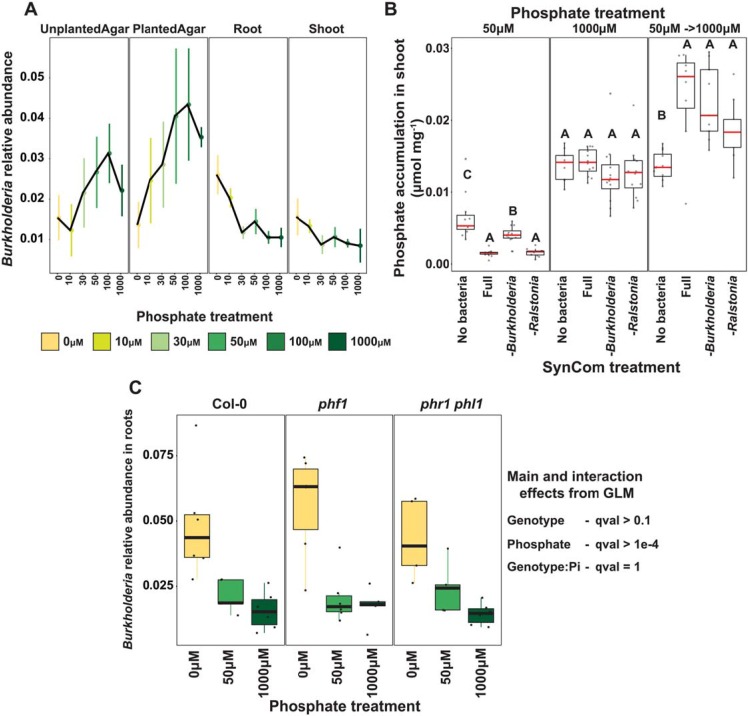
Phosphate starvation response (PSR) in nonmycorrhizal plants comprises transcriptional reprogramming resulting in severe physiological changes to the roots and shoots and repression of plant immunity. Thus, plant-colonizing microorganisms-the plant microbiota-are exposed to direct influence by the soil's phosphorus (P) content itself as well as to the indirect effects of soil P on the microbial niches shaped by the plant. The individual contribution of these factors to plant microbiota assembly remains unknown. To disentangle these direct and indirect effects, we planted PSR-deficient Arabidopsis mutants in a long-term managed soil P gradient and compared the composition of their shoot and root microbiota to wild-type plants across different P concentrations. PSR-deficiency had a larger effect on the composition of both bacterial and fungal plant-associated microbiota than soil P concentrations in both roots and shoots. To dissect plant-microbe interactions under variable P conditions, we conducted a microbiota reconstitution experiment. Using a 185-member bacterial synthetic community (SynCom) across a wide P concentration gradient in an agar matrix, we demonstrated a shift in the effect of bacteria on the plant from a neutral or positive interaction to a negative one, as measured by rosette size. This phenotypic shift was accompanied by changes in microbiota composition: the genus Burkholderia was specifically enriched in plant tissue under P starvation. Through a community drop-out experiment, we demonstrated that in the absence of Burkholderia from the SynCom, plant shoots accumulated higher ortophosphate (Pi) levels than shoots colonized with the full SynCom but only under Pi starvation conditions. Therefore, Pi-stressed plants are susceptible to colonization by latent opportunistic competitors found within their microbiome, thus exacerbating the plant's Pi starvation.
Conflict of interest statement
The authors have declared that no competing interests exist
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
