

A distinct class of eukaryotic MT-A70 methyltransferases maintain symmetric DNA N6-adenine methylation at the ApT dinucleotides as an epigenetic mark associated with transcription
- PMID: 31722409
- PMCID: PMC7145601
- DOI: 10.1093/nar/gkz1053
A distinct class of eukaryotic MT-A70 methyltransferases maintain symmetric DNA N6-adenine methylation at the ApT dinucleotides as an epigenetic mark associated with transcription
Abstract
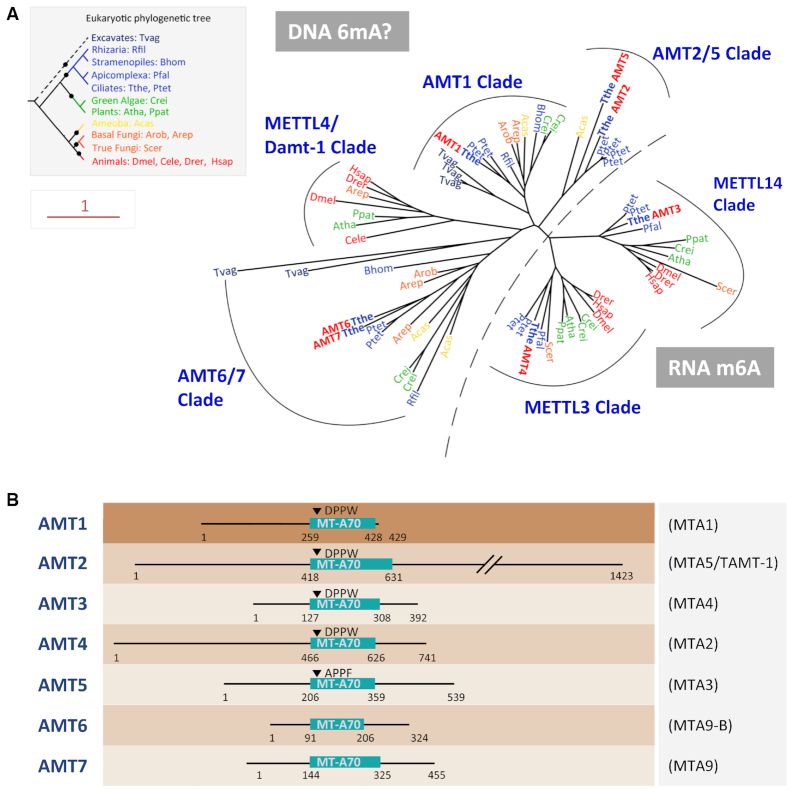
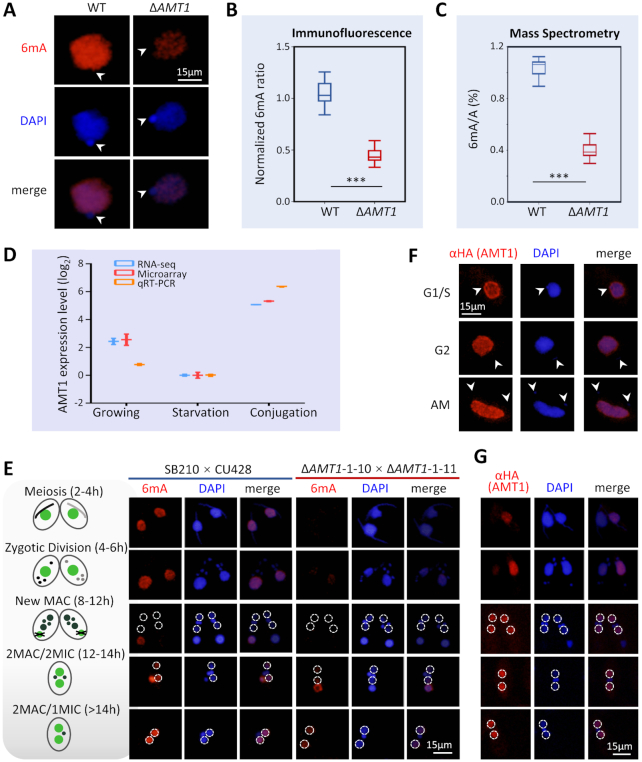
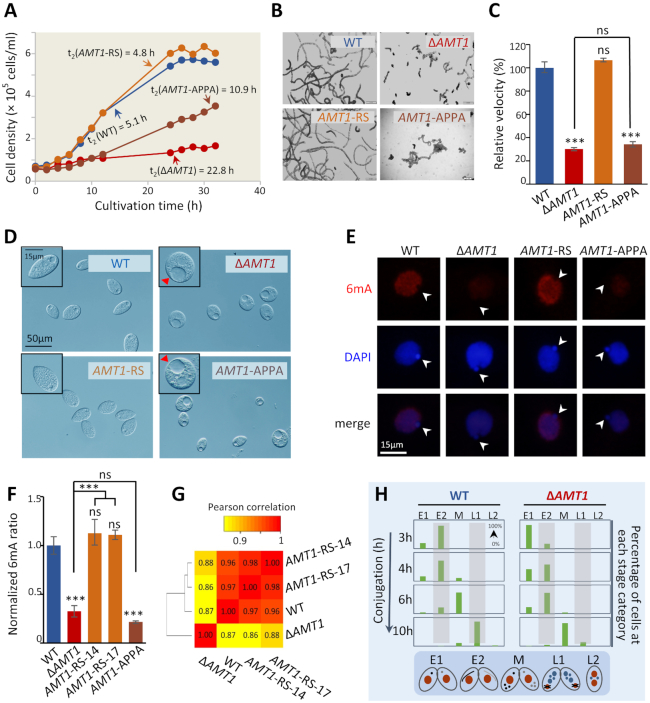
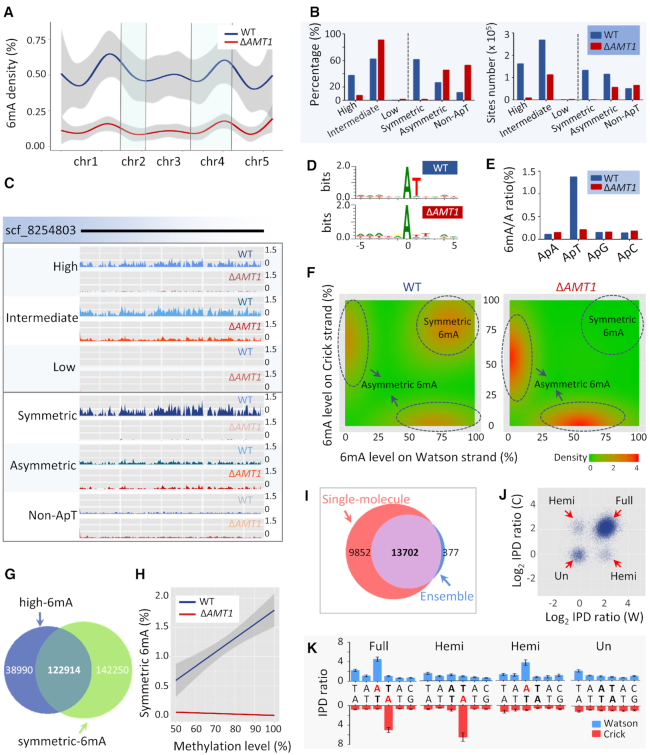
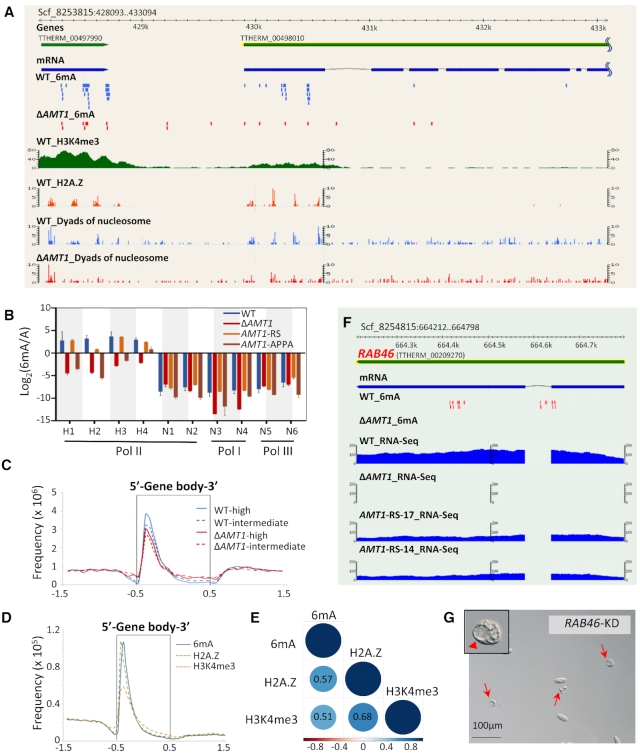
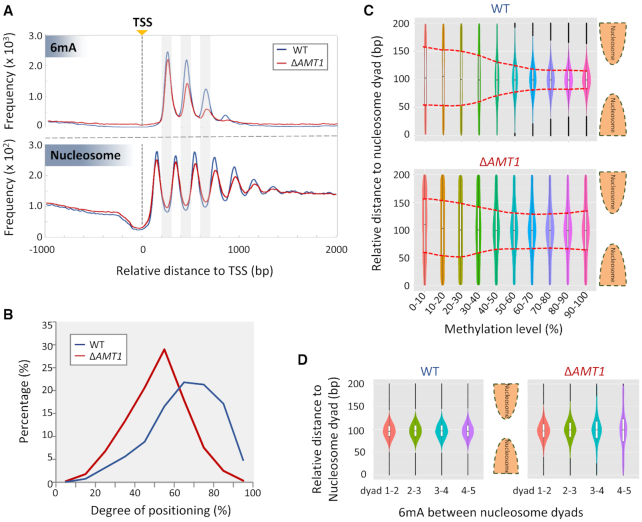
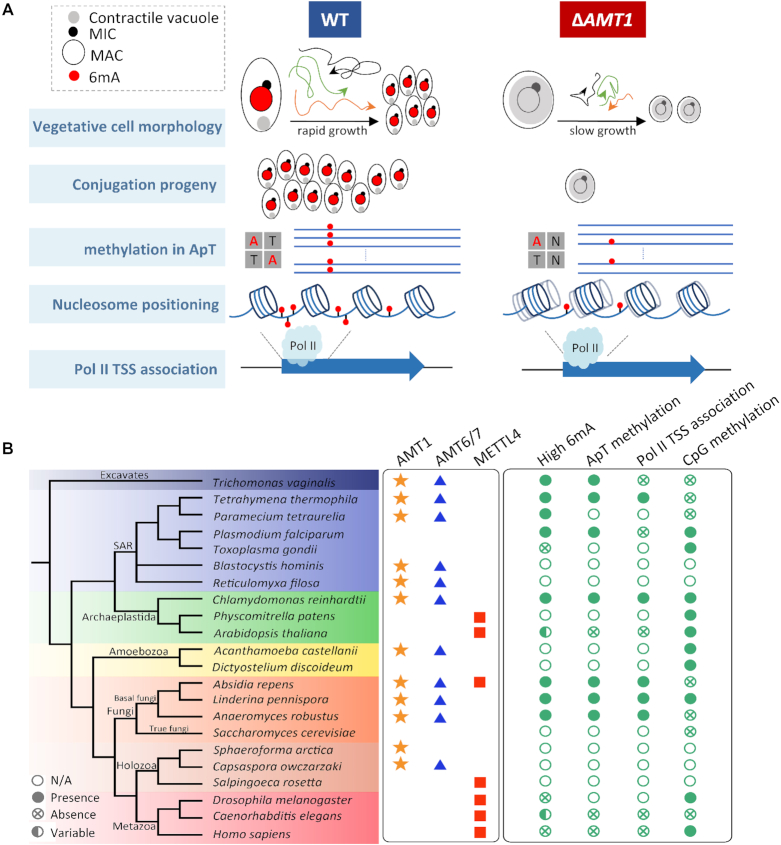
Rediscovered as a potential eukaryotic epigenetic mark, DNA N6-adenine methylation (6mA) varies across species in abundance and its relationships with transcription. Here we characterize AMT1-representing a distinct MT-A70 family methyltransferase-in the ciliate Tetrahymena thermophila. AMT1 loss-of-function leads to severe defects in growth and development. Single Molecule, Real-Time (SMRT) sequencing reveals that AMT1 is required for the bulk of 6mA and all symmetric methylation at the ApT dinucleotides. The detection of hemi-methylated ApT sites suggests a semi-conservative mechanism for maintaining symmetric methylation. AMT1 affects expression of many genes; in particular, RAB46, encoding a Rab family GTPase involved in contractile vacuole function, is likely a direct target. The distribution of 6mA resembles H3K4 methylation and H2A.Z, two conserved epigenetic marks associated with RNA polymerase II transcription. Furthermore, strong 6mA and nucleosome positioning in wild-type cells is attenuated in ΔAMT1 cells. Our results support that AMT1-catalyzed 6mA is an integral part of the transcription-associated epigenetic landscape. AMT1 homologues are generally found in protists and basal fungi featuring ApT hyper-methylation associated with transcription, which are missing in animals, plants, and true fungi. This dichotomy of 6mA functions and the underlying molecular mechanisms may have implications in eukaryotic diversification.
© The Author(s) 2019. Published by Oxford University Press on behalf of Nucleic Acids Research.
Figures
References
-
- Ammermann D., Steinbrück G., Baur R., Wohlert H.. Methylated bases in the DNA of the ciliate Stylonychia mytilus. Eur. J. Cell Biol. 1981; 24:154–156. - PubMed
-
- Cummings D.J., Tait A., Goddard J.M.. Methylated bases in DNA from Paramecium aurelia. Biochim. Biophys. Acta Nucleic Acids Protein Synth. 1974; 374:1–11. - PubMed
-
- Rae P.M., Steele R.E.. Modified bases in the DNAs of unicellular eukaryotes: an examination of distributions and possible roles, with emphasis on hydroxymethyluracil in dinoflagellates. Biosystems. 1978; 10:37–53. - PubMed
-
- Salvini M., Barone E., Ronca S., Nobili R.. DNA methylation in vegetative and conjugating cells of a protozoan ciliate: Blepharisma japonicum. Dev. Genet. 1986; 7:149–158.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
