

A Sequence Distance Graph framework for genome assembly and analysis
- PMID: 31723420
- PMCID: PMC6833988
- DOI: 10.12688/f1000research.20233.1
A Sequence Distance Graph framework for genome assembly and analysis
Abstract
The Sequence Distance Graph (SDG) framework works with genome assembly graphs and raw data from paired, linked and long reads. It includes a simple deBruijn graph module, and can import graphs using the graphical fragment assembly (GFA) format. It also maps raw reads onto graphs, and provides a Python application programming interface (API) to navigate the graph, access the mapped and raw data and perform interactive or scripted analyses. Its complete workspace can be dumped to and loaded from disk, decoupling mapping from analysis and supporting multi-stage pipelines. We present the design and implementation of the framework, and example analyses scaffolding a short read graph with long reads, and navigating paths in a heterozygous graph for a simulated parent-offspring trio dataset. SDG is freely available under the MIT license at https://github.com/bioinfologics/sdg.
Keywords: Genome graph; genome assembly.
Copyright: © 2019 Yanes L et al.
Conflict of interest statement
No competing interests were disclosed.
Figures
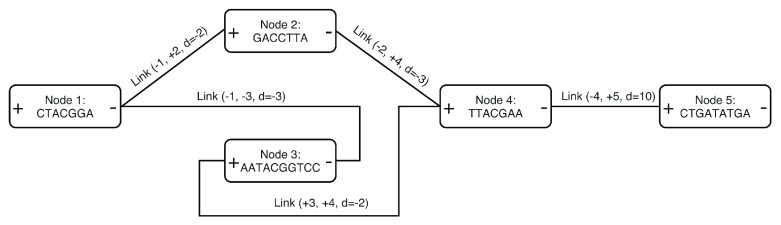
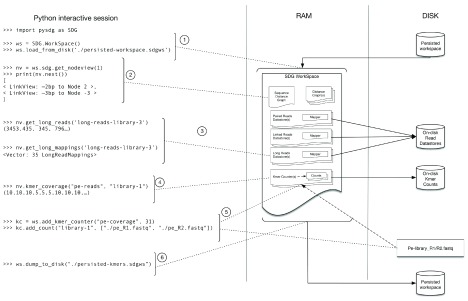

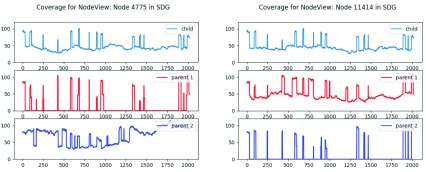
References
-
- Jackman SD, Myers EW, Gonella G: The GFA Specification. Reference Source
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources