

Cryo-EM structure of the complete and ligand-saturated insulin receptor ectodomain
- PMID: 31727777
- PMCID: PMC7039211
- DOI: 10.1083/jcb.201907210
Cryo-EM structure of the complete and ligand-saturated insulin receptor ectodomain
Abstract
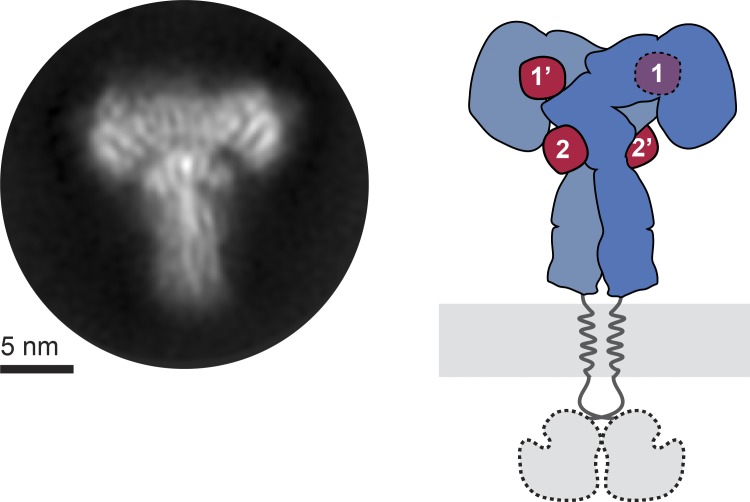
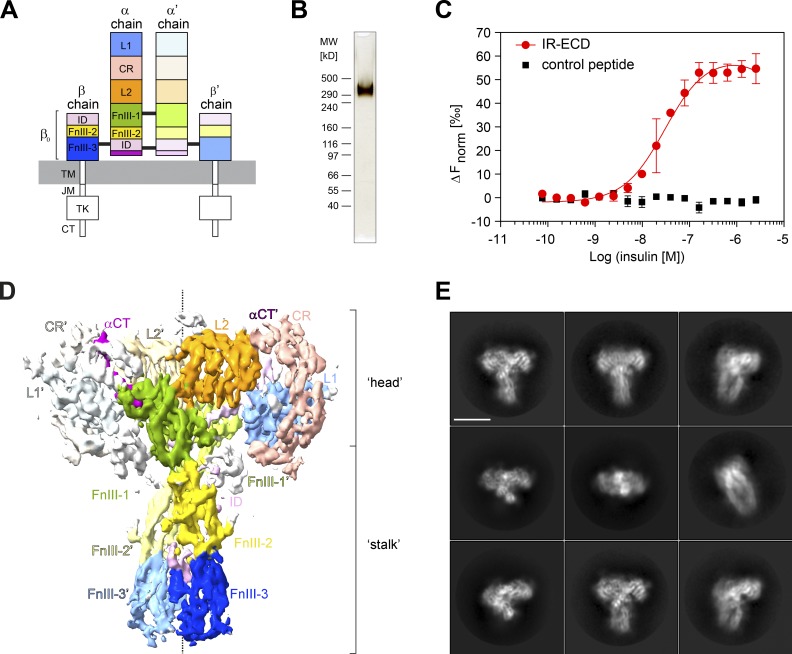
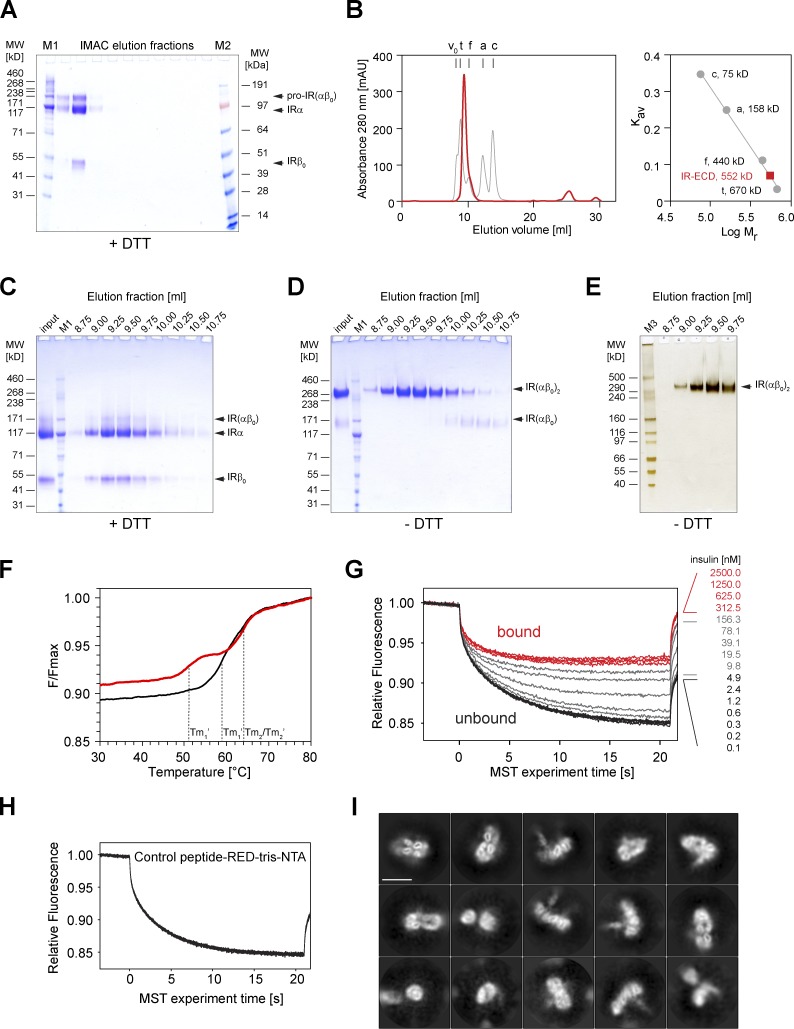
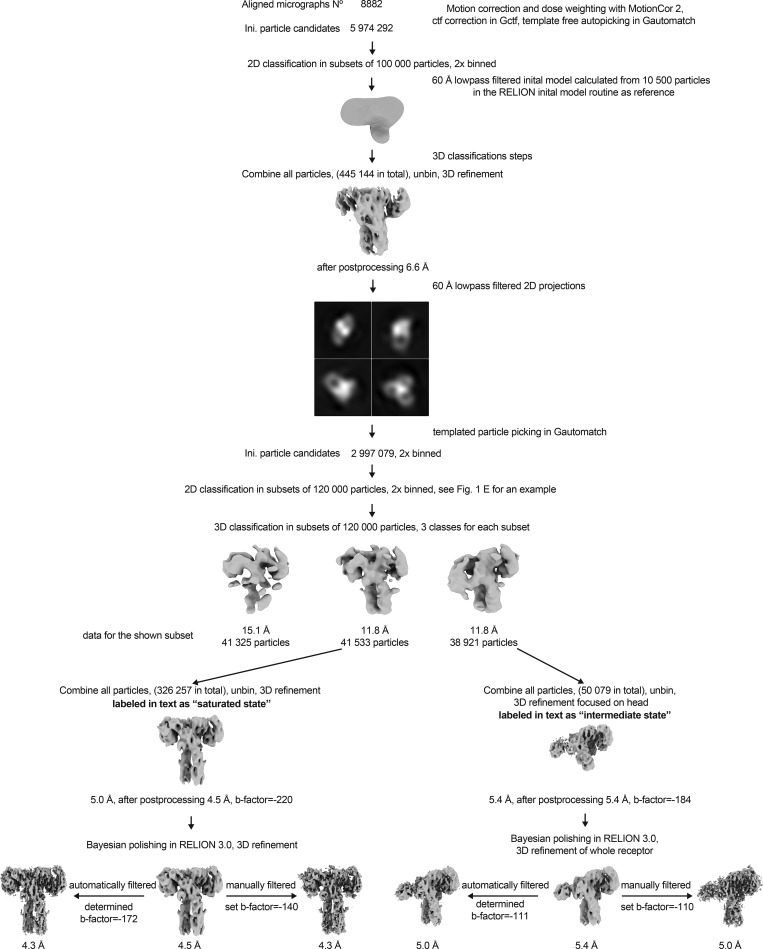
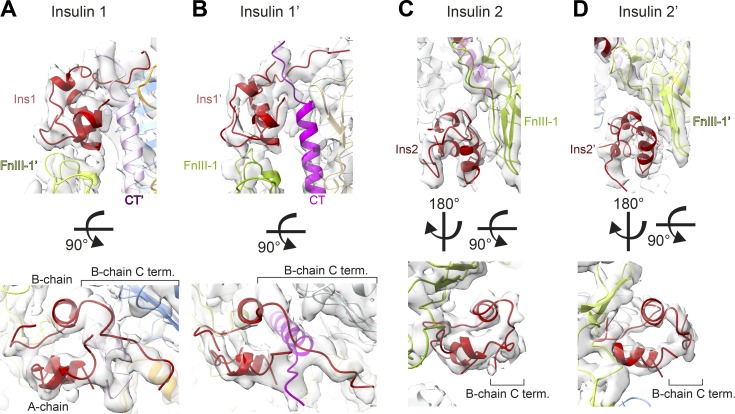
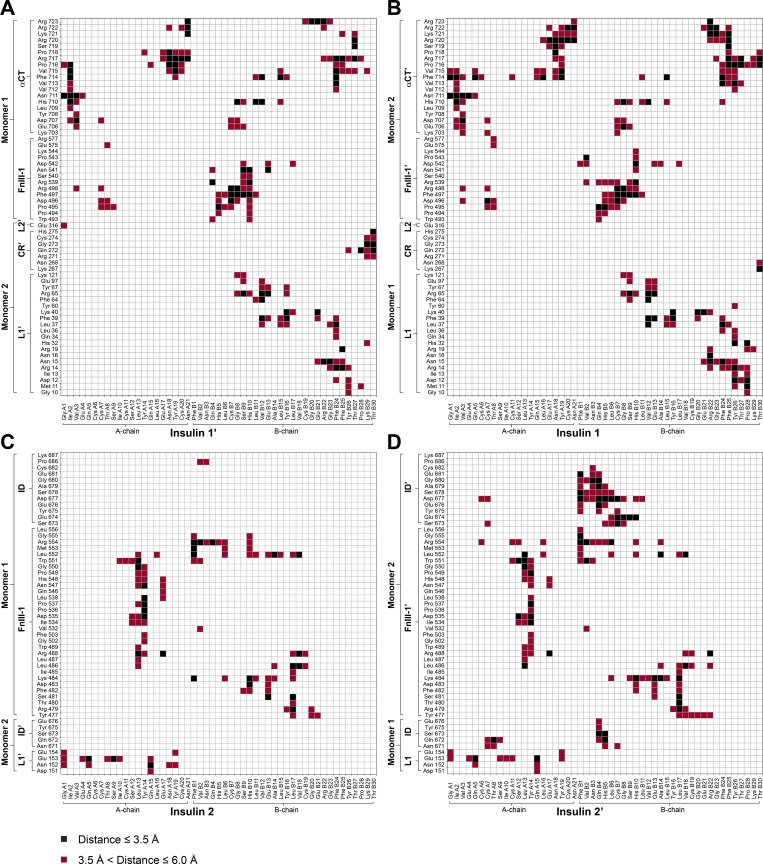
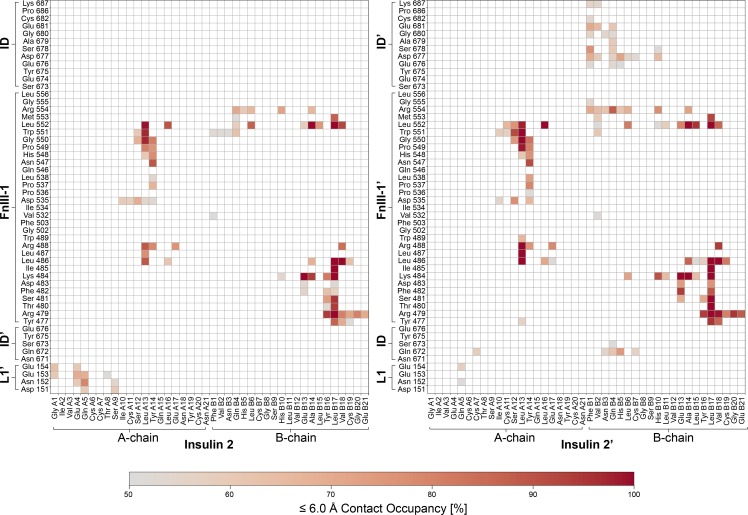
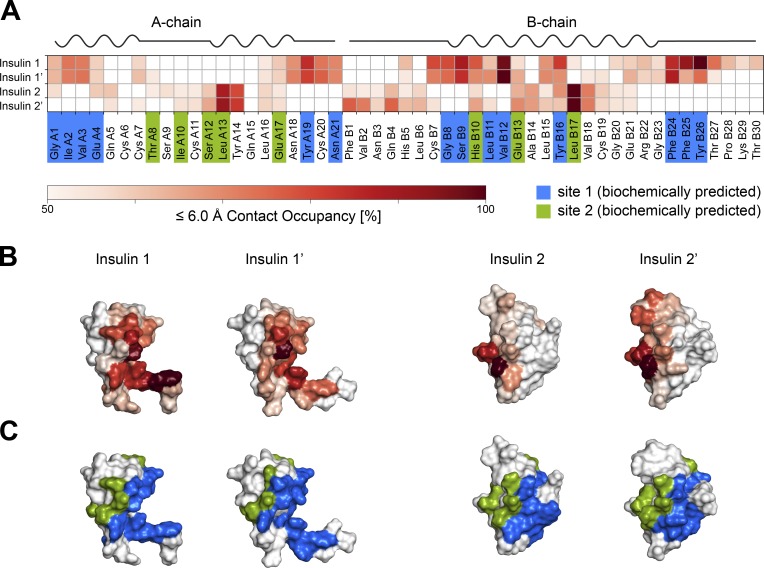
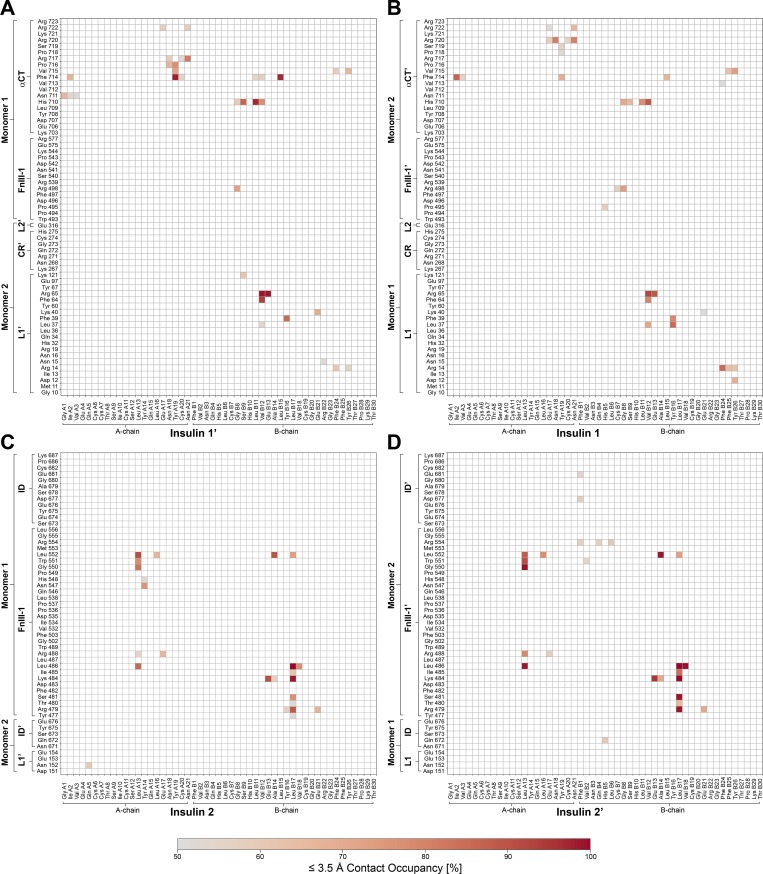
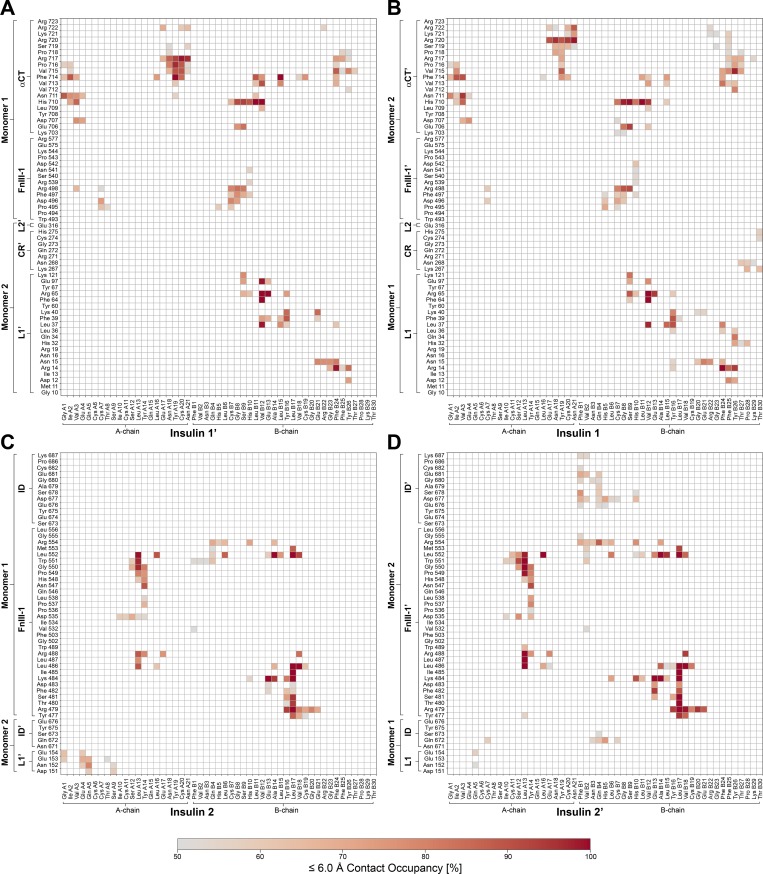
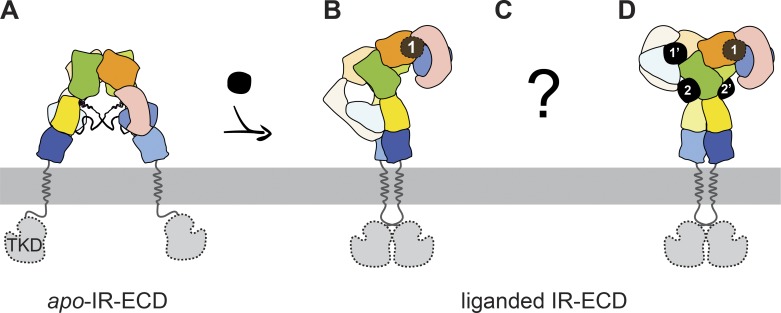
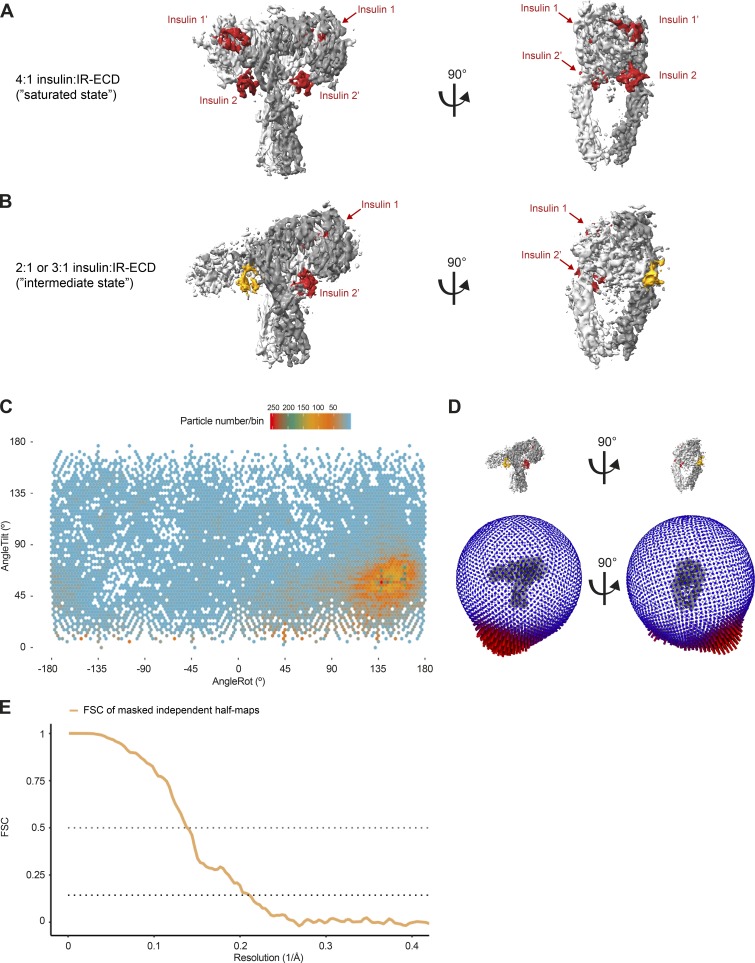
Glucose homeostasis and growth essentially depend on the hormone insulin engaging its receptor. Despite biochemical and structural advances, a fundamental contradiction has persisted in the current understanding of insulin ligand-receptor interactions. While biochemistry predicts two distinct insulin binding sites, 1 and 2, recent structural analyses have resolved only site 1. Using a combined approach of cryo-EM and atomistic molecular dynamics simulation, we present the structure of the entire dimeric insulin receptor ectodomain saturated with four insulin molecules. Complementing the previously described insulin-site 1 interaction, we present the first view of insulin bound to the discrete insulin receptor site 2. Insulin binding stabilizes the receptor ectodomain in a T-shaped conformation wherein the membrane-proximal domains converge and contact each other. These findings expand the current models of insulin binding to its receptor and of its regulation. In summary, we provide the structural basis for a comprehensive description of ligand-receptor interactions that ultimately will inform new approaches to structure-based drug design.
© 2019 Gutmann et al.
Figures
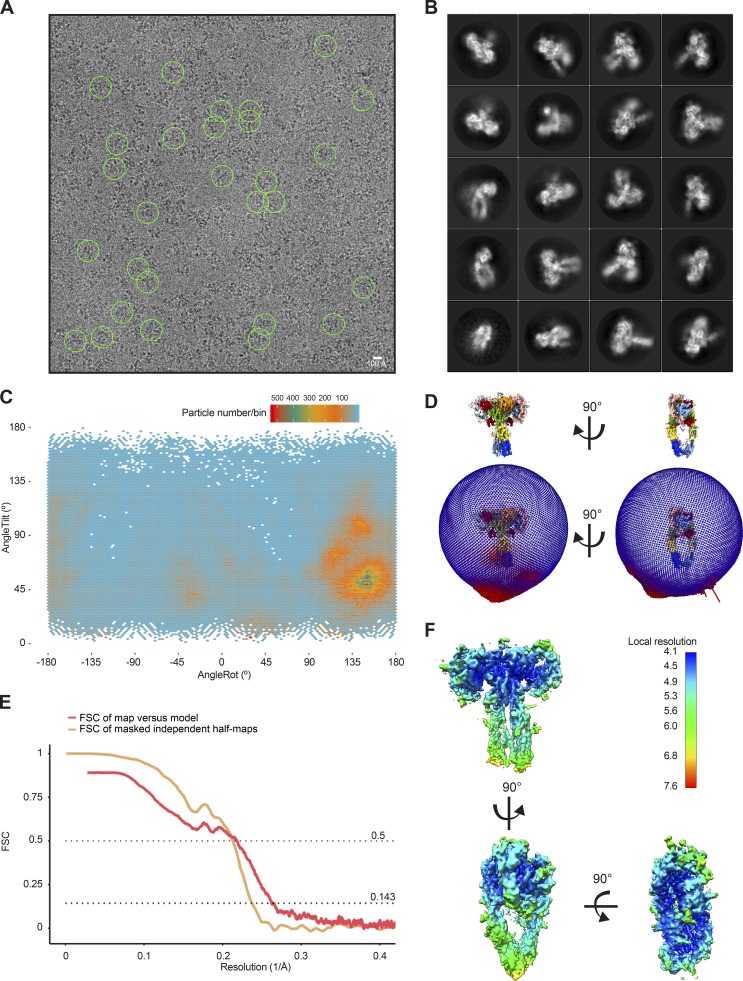
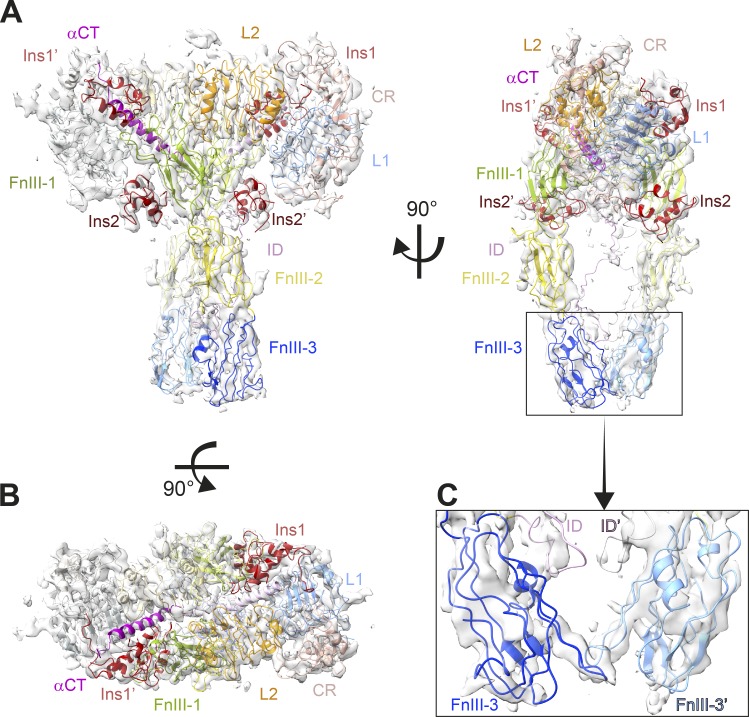
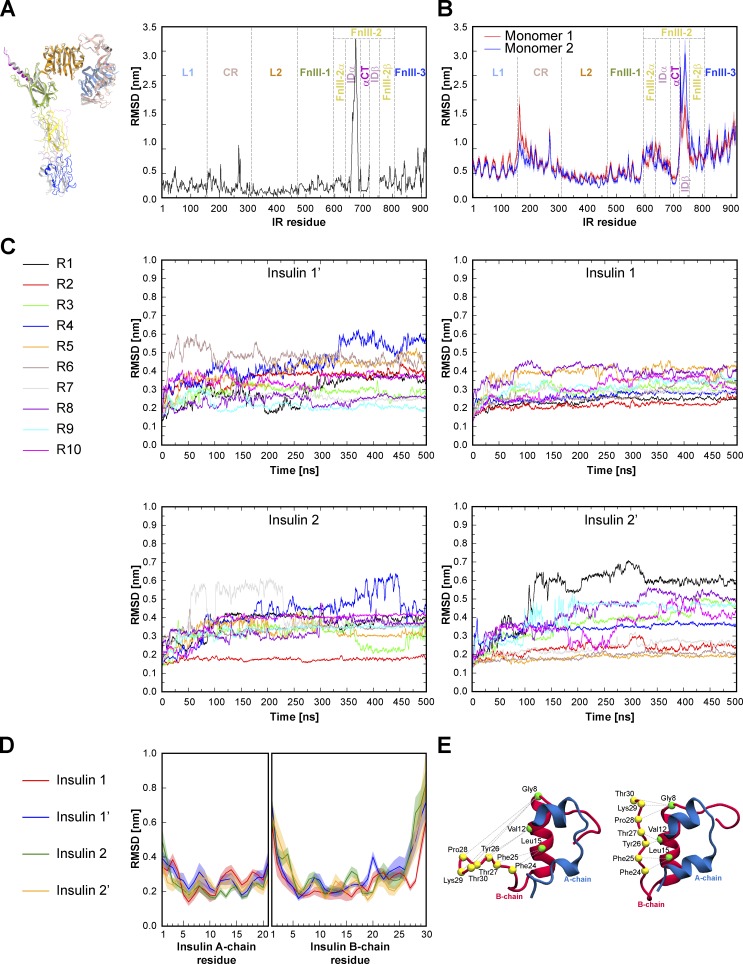
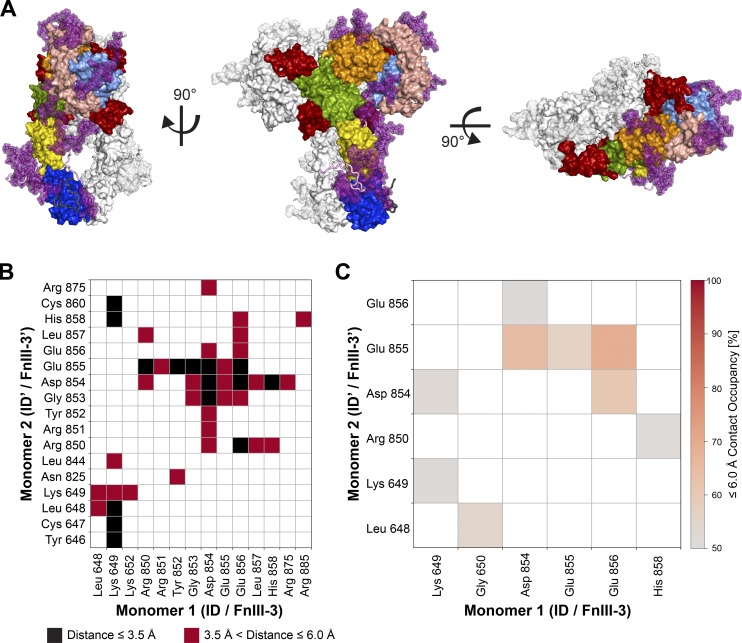
References
-
- Adams M.J., Blundell T.L., Dodson E.J., Dodson G.G., Vijayan M., Baker E.N., Harding M.M., Hodgkin D.C., Rimmer B., and Sheat S.. 1969. Structure of Rhombohedral 2 Zinc Insulin Crystals. Nature. 224:491-495.
-
- Baker E.N., Blundell T.L., Cutfield J.F., Cutfield S.M., Dodson E.J., Dodson G.G., Hodgkin D.M., Hubbard R.E., Isaacs N.W., Reynolds C.D., et al. . 1988. The structure of 2Zn pig insulin crystals at 1.5 A resolution. Philos. Trans. R. Soc. Lond. B Biol. Sci. 319:369–456. 10.1098/rstb.1988.0058 - DOI - PubMed
-
- Bartoschik T., Galinec S., Kleusch C., Walkiewicz K., Breitsprecher D., Weigert S., Muller Y.A., You C., Piehler J., Vercruysse T., et al. . 2018. Near-native, site-specific and purification-free protein labeling for quantitative protein interaction analysis by MicroScale Thermophoresis. Sci. Rep. 8:4977 10.1038/s41598-018-23154-3 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
