

Destabilization of NOXA mRNA as a common resistance mechanism to targeted therapies
- PMID: 31727958
- PMCID: PMC6856172
- DOI: 10.1038/s41467-019-12477-y
Destabilization of NOXA mRNA as a common resistance mechanism to targeted therapies
Abstract
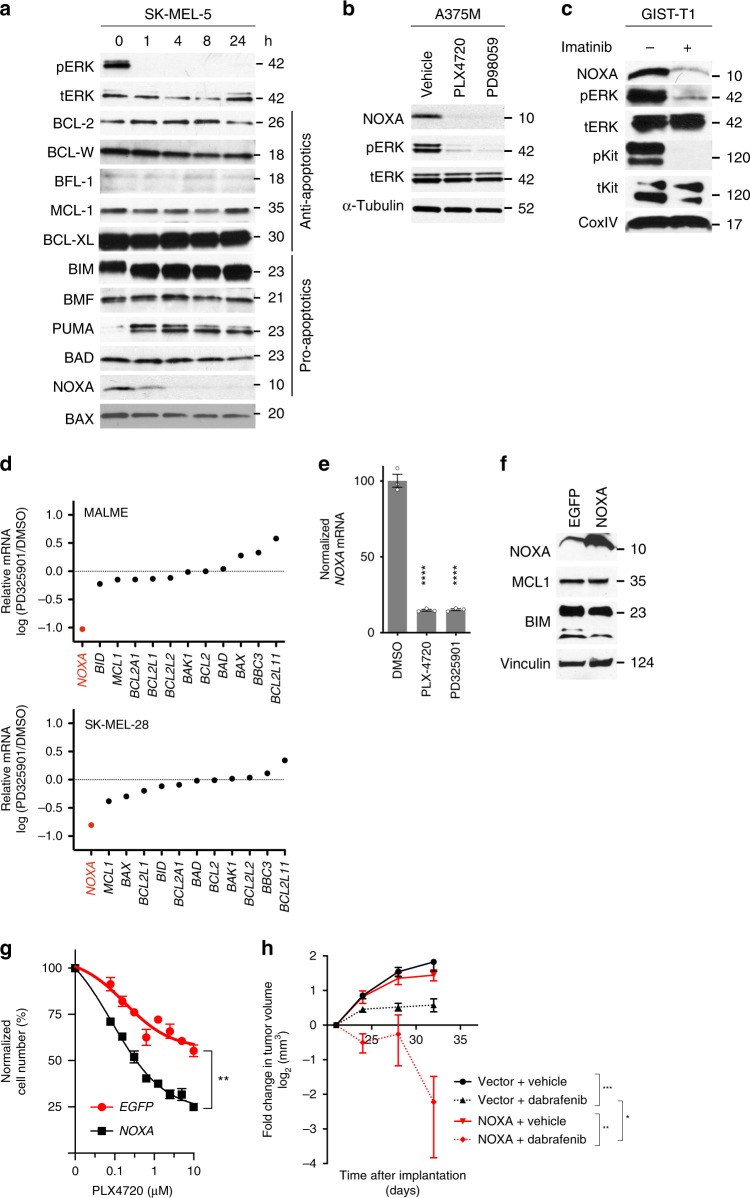
Most targeted cancer therapies fail to achieve complete tumor regressions or attain durable remissions. To understand why these treatments fail to induce robust cytotoxic responses despite appropriately targeting oncogenic drivers, here we systematically interrogated the dependence of cancer cells on the BCL-2 family of apoptotic proteins after drug treatment. We observe that multiple targeted therapies, including BRAF or EGFR inhibitors, rapidly deplete the pro-apoptotic factor NOXA, thus creating a dependence on the anti-apoptotic protein MCL-1. This adaptation requires a pathway leading to destabilization of the NOXA mRNA transcript. We find that interruption of this mechanism of anti-apoptotic adaptive resistance dramatically increases cytotoxic responses in cell lines and a murine melanoma model. These results identify NOXA mRNA destabilization/MCL-1 adaptation as a non-genomic mechanism that limits apoptotic responses, suggesting that sequencing of MCL-1 inhibitors with targeted therapies could overcome such widespread and clinically important resistance.
Conflict of interest statement
K.F. is a member of the board of directors at Loxo Oncology, Clovis Oncology, Strata Oncology, Vivid Biosciences; the corporate advisory board of X4 Pharmaceuticals, PIC Therapeutics; the scientific advisory board of Sanofi, Amgen, Asana, Adaptimmune, Fount, Aeglea, Array BioPharma, Shattuck Labs, Arch Oncology, Tolero, Apricity, Oncoceutics, Fog Pharma, Tvardi; and consultant for Novartis, Genentech, Bristol-Myers Squibb, Merck, Takeda, Verastem, Checkmate, Boston Biomedical. F.S.H. is a consultant for Bristol-Myers Squibb, Merck, EMD Serono, Novartis, Celldex, Amgen, Genetech, Incyte, Bayer, Aduro, Partners Therapeutics, Sanofi, Pfizer, Pionyr; member of scientific advisory board of Apricity; member of the advisory board of Pionyr, 7 Hills Pharma, Verastem. R.H. has received research grants from Bristol-Myers-Squibb and Novartis. J.R.C., A.E.T., and J.P.S. are employees of Astra-Zeneca. C.Y. is a consultant to Merck. J.M. is a consultant for Vivid Biosciences and Oncoheroes Biosciences. D.E.F. has a financial interest in Soltego, Inc., a company developing SIK inhibitors for topical skin darkening treatments that might be used for a broad set of human applications. These interests were reviewed and are managed by Massachusetts General Hospital and Partners HealthCare in accordance with their conflict of interest policies. The remaining authors declare no competing interests.
Figures




Comment in
-
Overcoming MCL-1-driven adaptive resistance to targeted therapies.Nat Commun. 2020 Jan 27;11(1):531. doi: 10.1038/s41467-020-14392-z. Nat Commun. 2020. PMID: 31988312 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
