

Periodic Oxaliplatin Administration in Synergy with PER2-Mediated PCNA Transcription Repression Promotes Chronochemotherapeutic Efficacy of OSCC
- PMID: 31728273
- PMCID: PMC6839751
- DOI: 10.1002/advs.201900667
Periodic Oxaliplatin Administration in Synergy with PER2-Mediated PCNA Transcription Repression Promotes Chronochemotherapeutic Efficacy of OSCC
Abstract
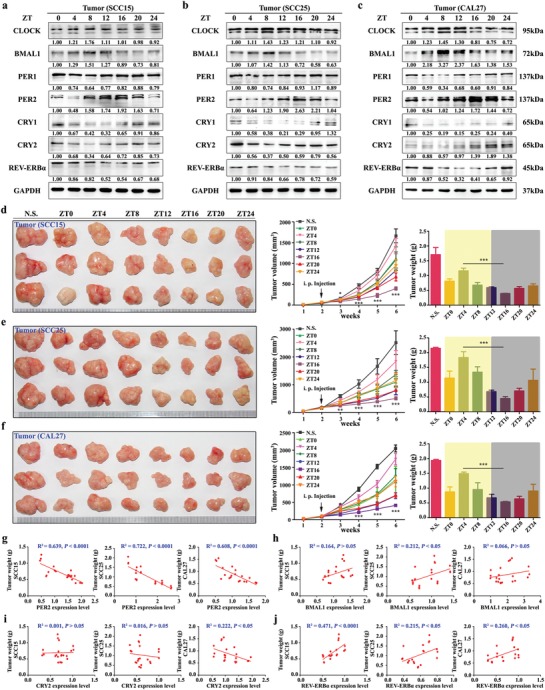
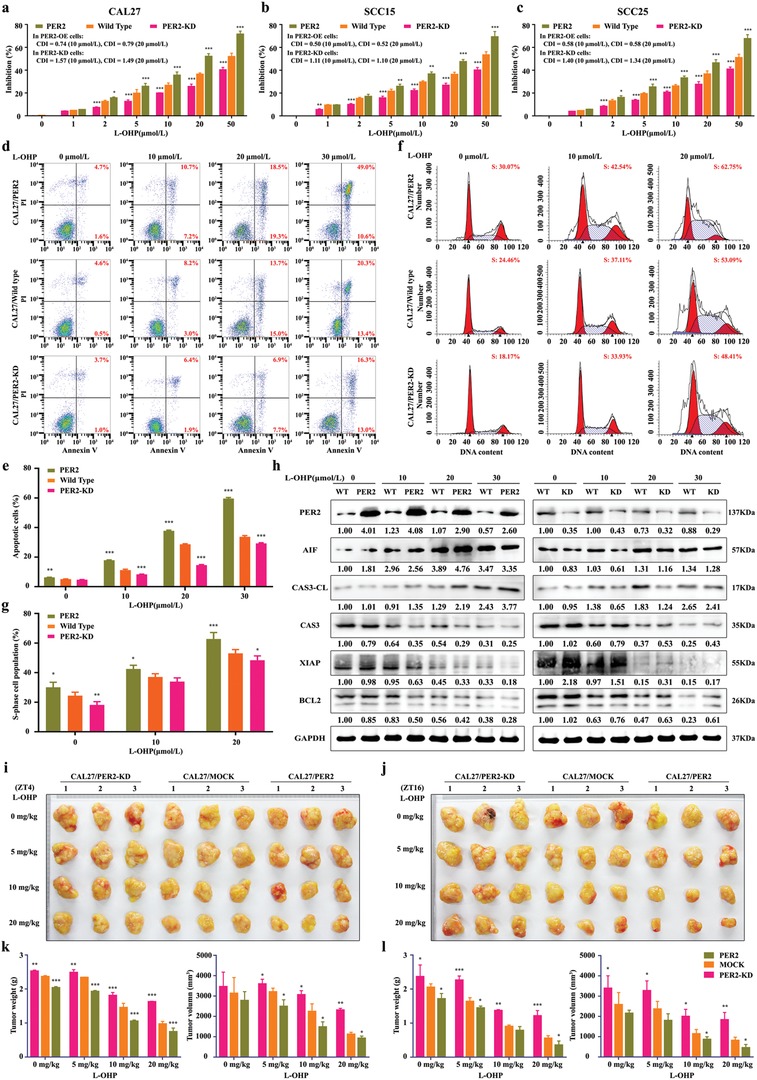
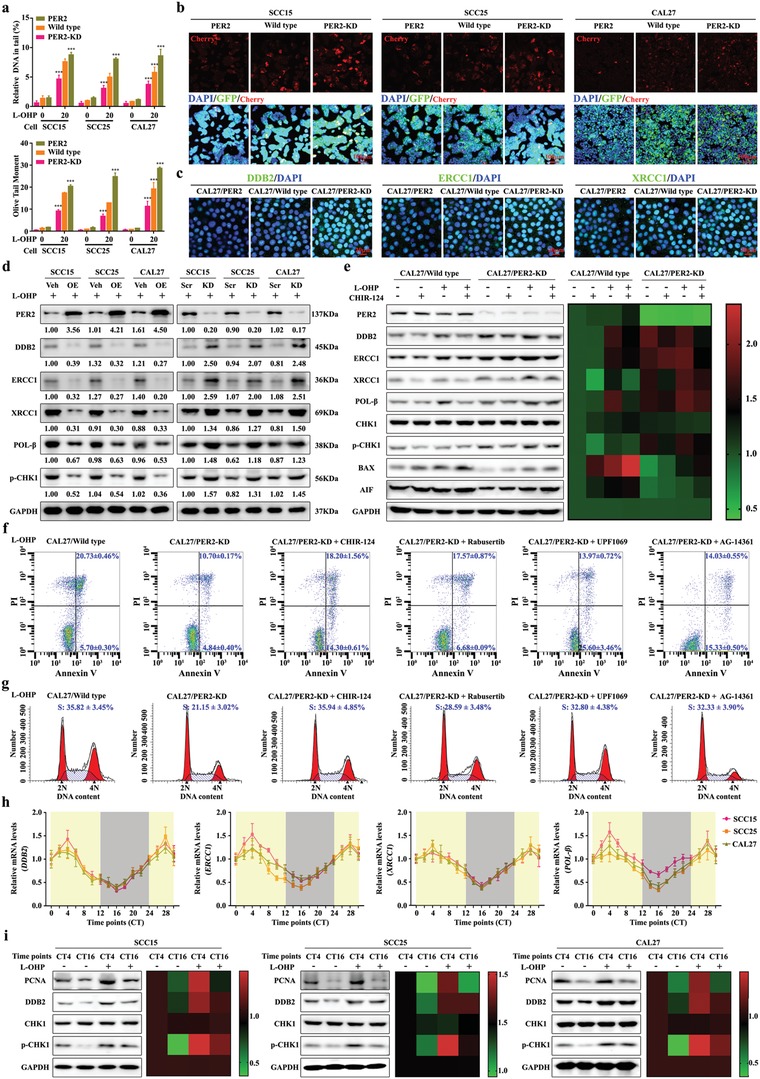
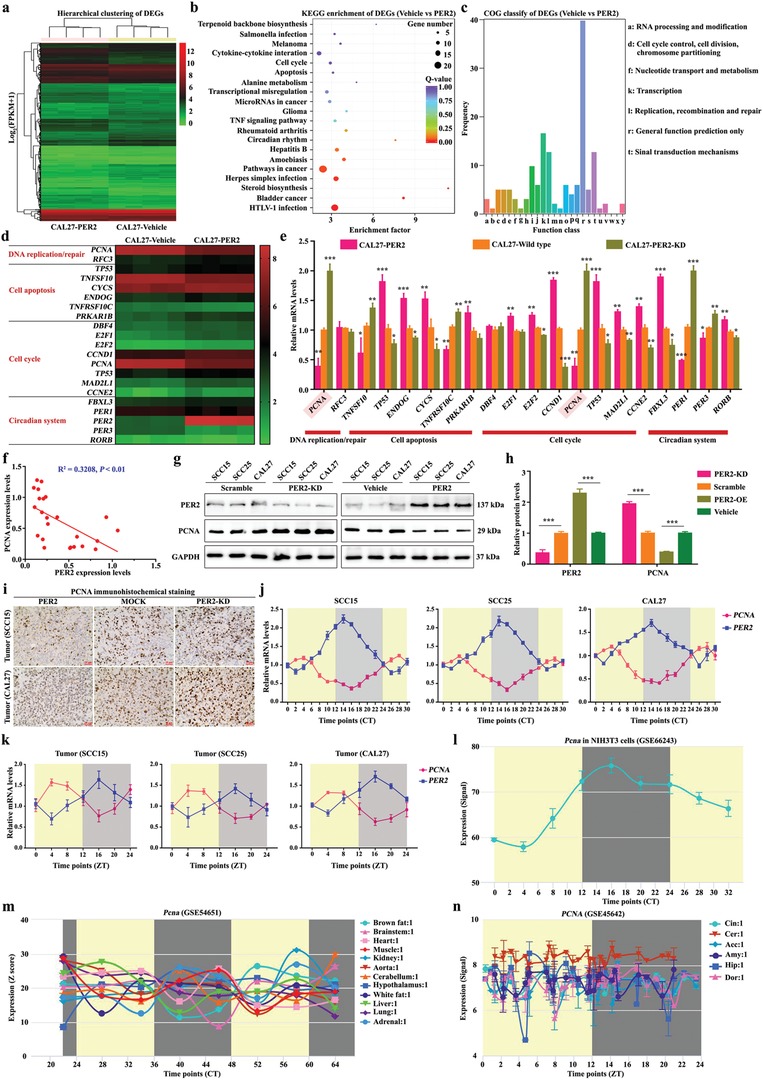
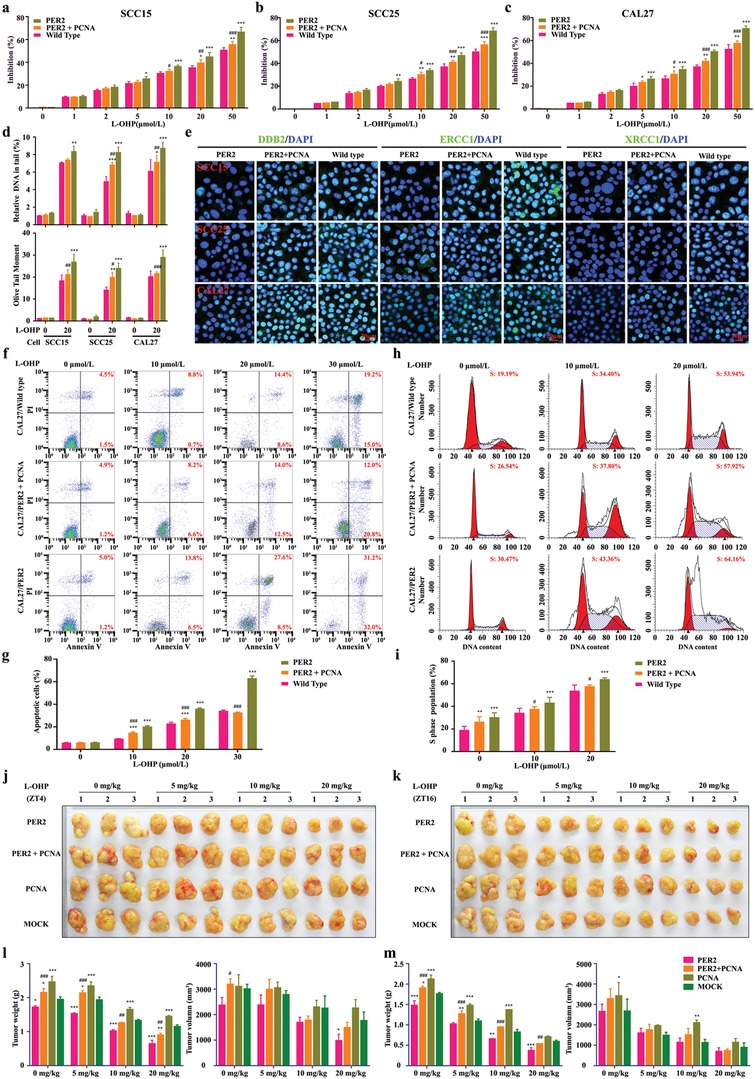
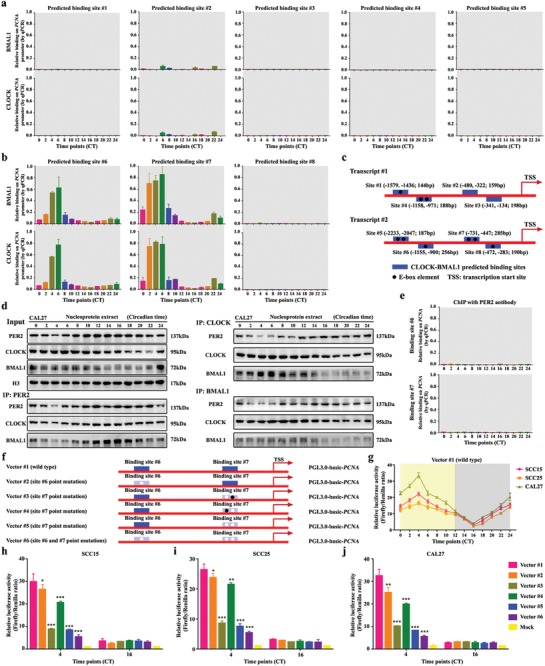
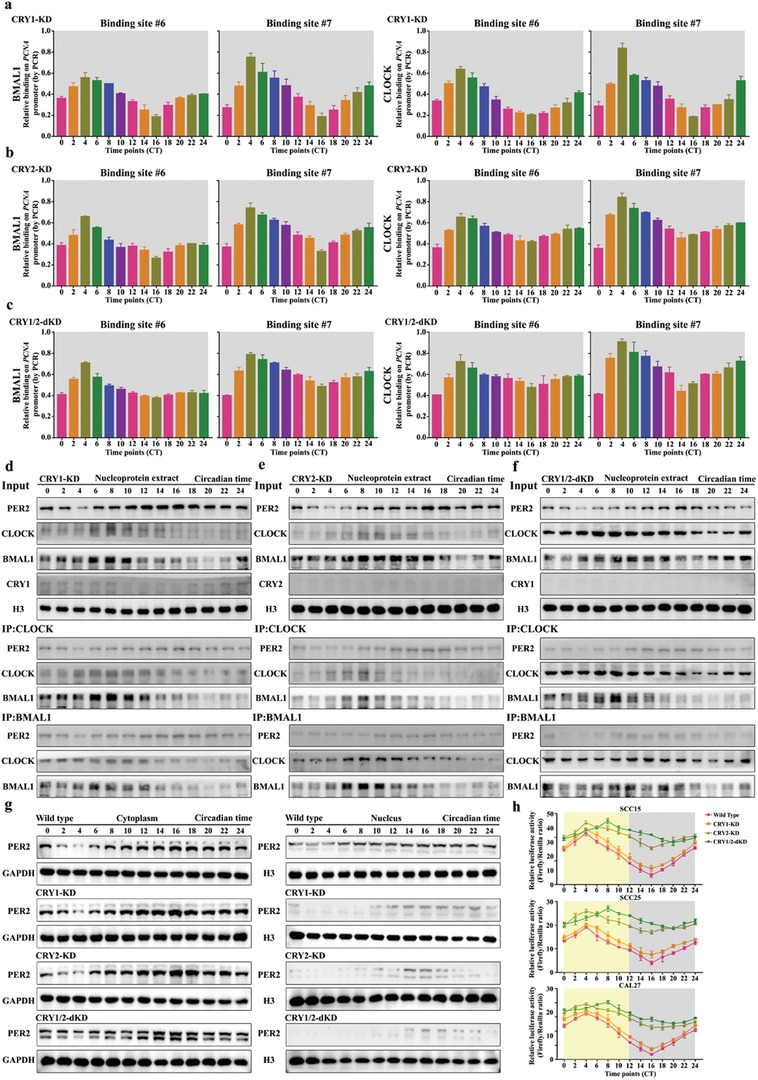
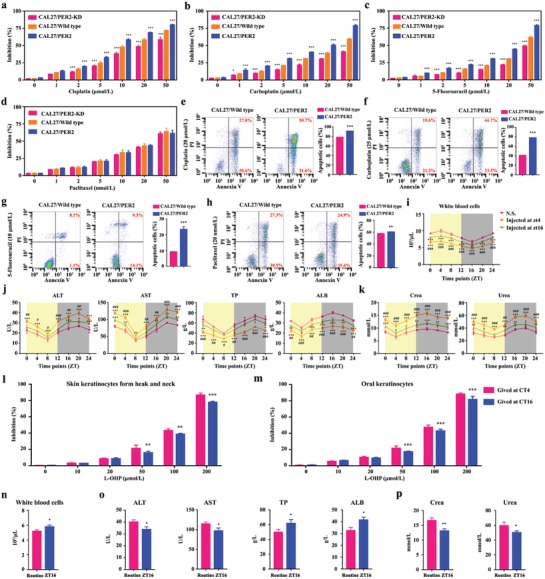
Developing chemotherapeutic resistance affects clinical outcomes of oxaliplatin treatment on various types of cancer. Thus, it is imperative to explore alternative therapeutic strategies to improve the efficacy of oxaliplatin. Here, it is shown that circadian regulator period 2 (PER2) can potentiate the cytotoxicity of oxaliplatin and boost cell apoptosis by inhibiting DNA adducts repair in human oral squamous cell carcinoma (OSCC) cells. The circadian timing system is closely involved in controling the activity of DNA adducts repair and gives it a 24 h rhythm. The mechanistic dissection clarifies that PER2 can periodically suppress proliferating cell nuclear antigen (PCNA) transcription by pulling down circadian locomotor output cycles kaput-brain and muscle arnt-like 1 heterodimer from PCNA promoter in a CRY1/2-dependent manner, which subsequently impedes oxaliplatin-induced DNA adducts repair. Similarly, PER2 is capable of improving the efficacy of classical DNA-damaging chemotherapeutic agents. The tumor-bearing mouse model displays PER2 can be deployed as an oxaliplatin administration timing biomarker. In summary, it is believed that the chronochemotherapeutic strategy matching PER2 expression rhythm can efficiently improve the oxaliplatin efficacy of OSCC.
Keywords: DNA‐damaging repair; chronochemotherapeutic strategy; circadian clock genes; oral squamous cell carcinoma; oxaliplatin.
© 2019 The Authors. Published by WILEY‐VCH Verlag GmbH & Co. KGaA, Weinheim.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- a) Torre L. A., Bray F., Siegel R. L., Ferlay J., Lortet‐Tieulent J., Jemal A., Ca ‐ Cancer J. Clin. 2015, 65, 87; - PubMed
- b) Carnielli C. M., Macedo C. C. S., De Rossi T., Granato D. C., Rivera C., Domingues R. R., Pauletti B. A., Yokoo S., Heberle H., Busso‐Lopes A. F., Cervigne N. K., Sawazaki‐Calone I., Meirelles G. V., Marchi F. A., Telles G. P., Minghim R., Ribeiro A. C. P., Brandao T. B., de Castro G. Jr., Gonzalez‐Arriagada W. A., Gomes A., Penteado F., Santos‐Silva A. R., Lopes M. A., Rodrigues P. C., Sundquist E., Salo T., da Silva S. D., Alaoui‐Jamali M. A., Graner E., Fox J. W., Coletta R. D., Leme A. F. P., Nat. Commun. 2018, 9, 3598. - PMC - PubMed
-
- Raymond E., Chaney S. G., Taamma A., Cvitkovic E., Ann. Oncol. 1998, 9, 1053. - PubMed
-
- Pignon J. P., Bourhis J., Domenge C., Designe L., Lancet 2000, 355, 949. - PubMed
LinkOut - more resources
Full Text Sources
Miscellaneous