

Unifying machine learning and quantum chemistry with a deep neural network for molecular wavefunctions
- PMID: 31729373
- PMCID: PMC6858523
- DOI: 10.1038/s41467-019-12875-2
Unifying machine learning and quantum chemistry with a deep neural network for molecular wavefunctions
Abstract
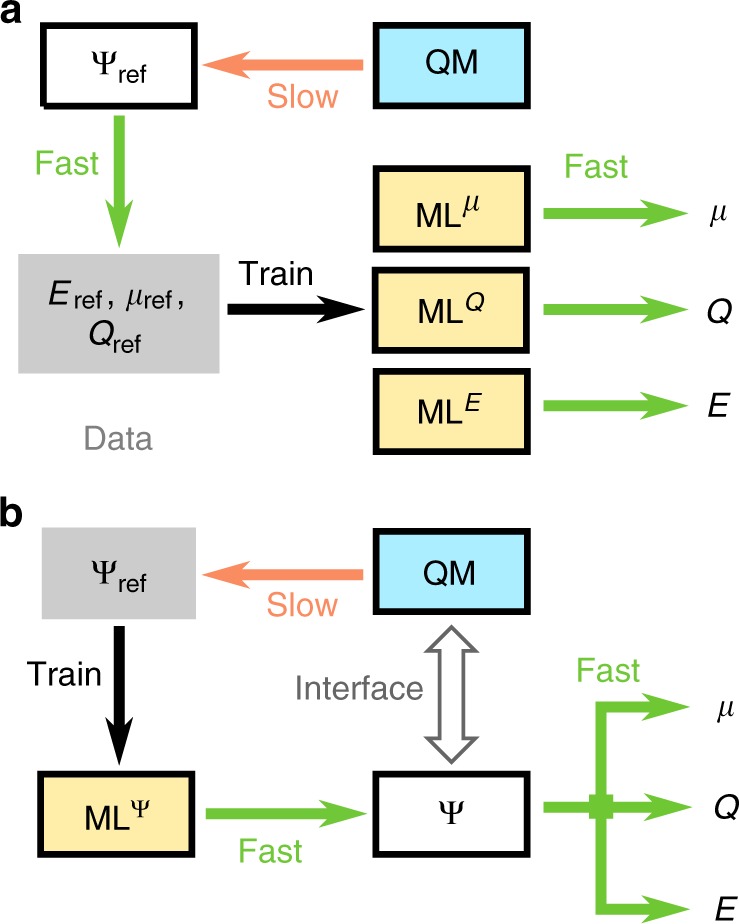
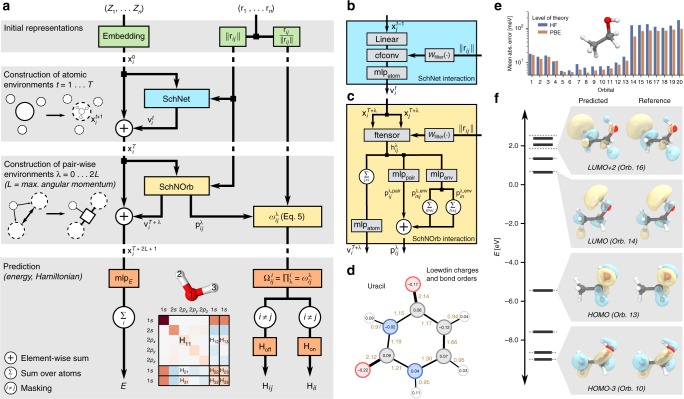
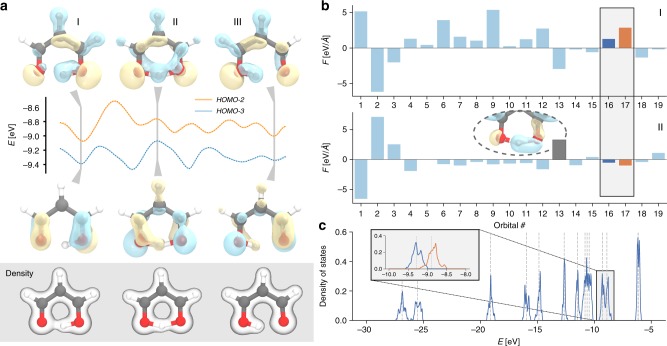
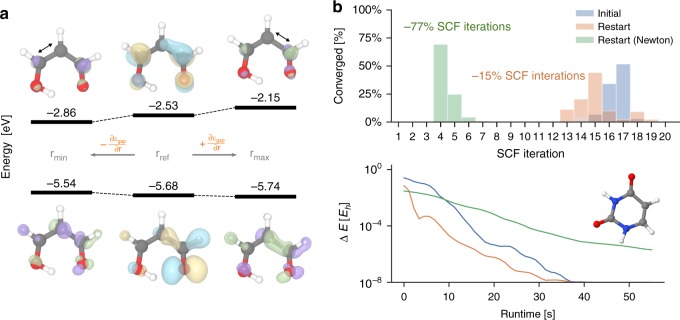
Machine learning advances chemistry and materials science by enabling large-scale exploration of chemical space based on quantum chemical calculations. While these models supply fast and accurate predictions of atomistic chemical properties, they do not explicitly capture the electronic degrees of freedom of a molecule, which limits their applicability for reactive chemistry and chemical analysis. Here we present a deep learning framework for the prediction of the quantum mechanical wavefunction in a local basis of atomic orbitals from which all other ground-state properties can be derived. This approach retains full access to the electronic structure via the wavefunction at force-field-like efficiency and captures quantum mechanics in an analytically differentiable representation. On several examples, we demonstrate that this opens promising avenues to perform inverse design of molecular structures for targeting electronic property optimisation and a clear path towards increased synergy of machine learning and quantum chemistry.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Podryabinkin EV, Shapeev AV. Active learning of linearly parametrized interatomic potentials. Comput. Mater. Sci. 2017;140:171–180. doi: 10.1016/j.commatsci.2017.08.031. - DOI
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
