

Structural variation in the sequencing era
- PMID: 31729472
- PMCID: PMC7402362
- DOI: 10.1038/s41576-019-0180-9
Structural variation in the sequencing era
Abstract
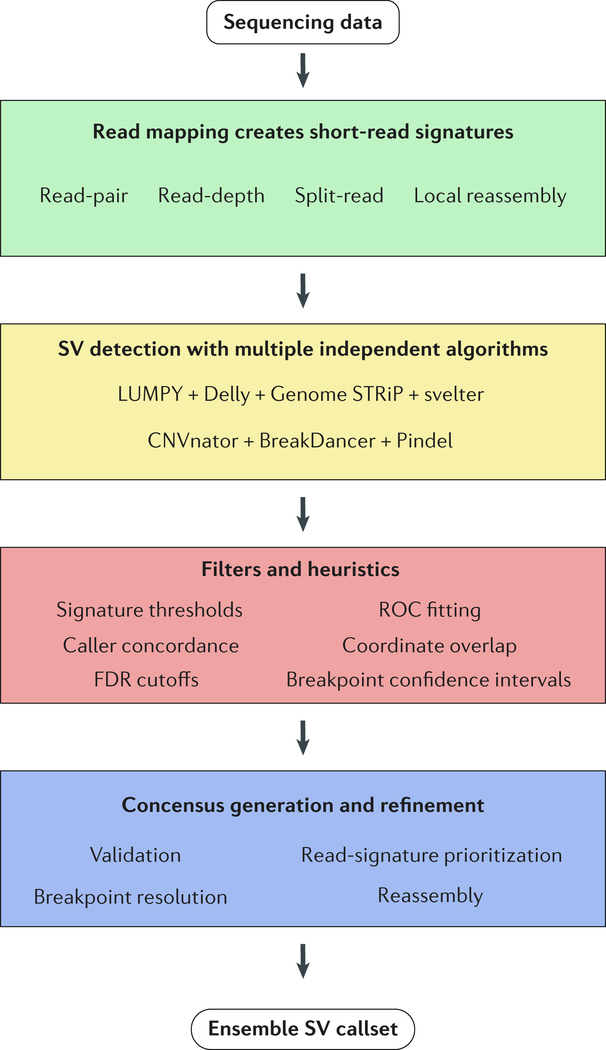
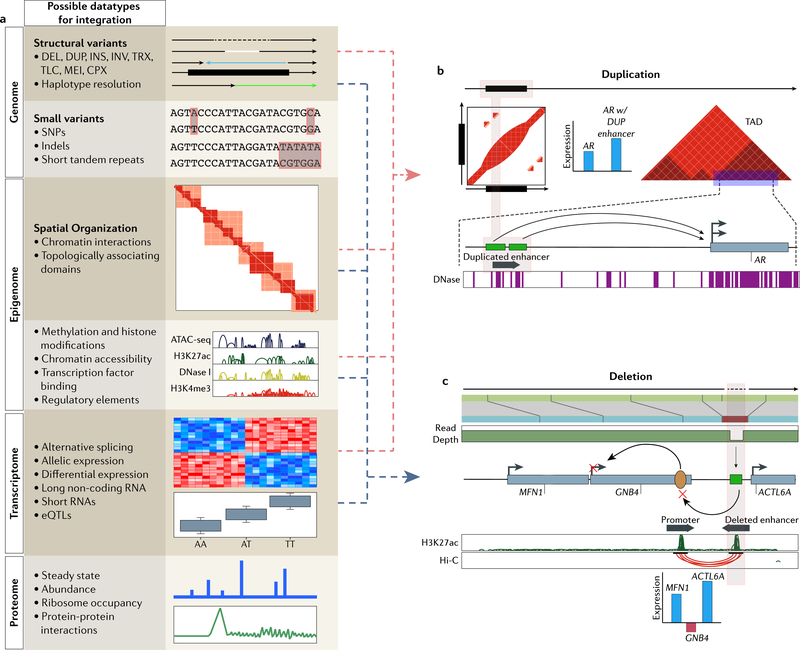
Identifying structural variation (SV) is essential for genome interpretation but has been historically difficult due to limitations inherent to available genome technologies. Detection methods that use ensemble algorithms and emerging sequencing technologies have enabled the discovery of thousands of SVs, uncovering information about their ubiquity, relationship to disease and possible effects on biological mechanisms. Given the variability in SV type and size, along with unique detection biases of emerging genomic platforms, multiplatform discovery is necessary to resolve the full spectrum of variation. Here, we review modern approaches for investigating SVs and proffer that, moving forwards, studies integrating biological information with detection will be necessary to comprehensively understand the impact of SV in the human genome.
Conflict of interest statement
Competing interests
The authors declare no competing interests.
Figures

References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
