

Gene Expression Changes and Community Turnover Differentially Shape the Global Ocean Metatranscriptome
- PMID: 31730850
- PMCID: PMC6912165
- DOI: 10.1016/j.cell.2019.10.014
Gene Expression Changes and Community Turnover Differentially Shape the Global Ocean Metatranscriptome
Abstract
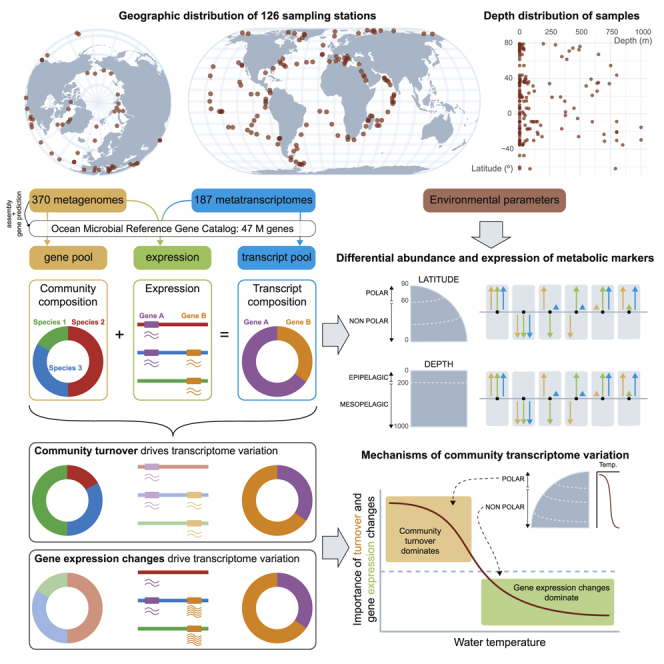
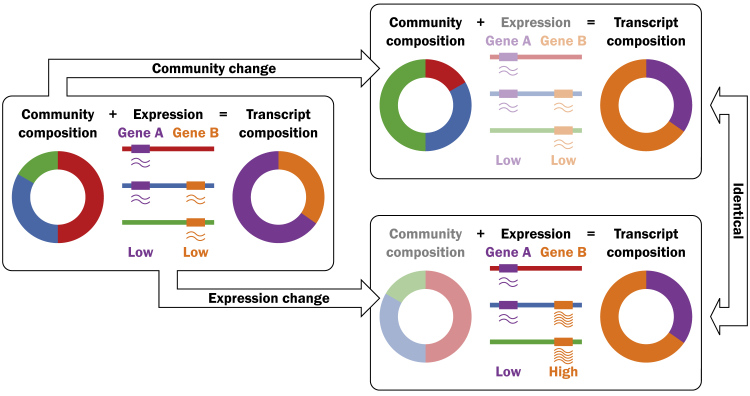
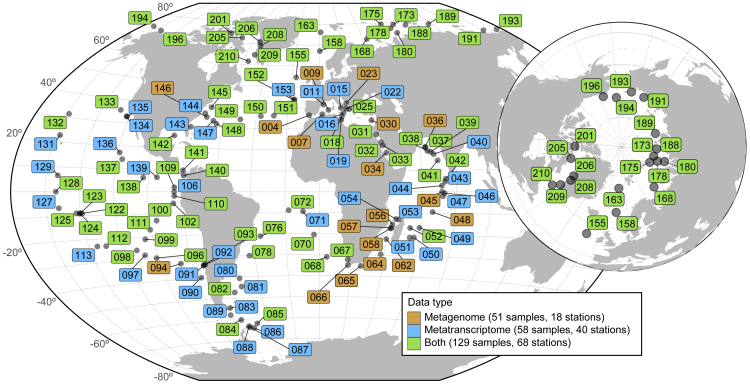
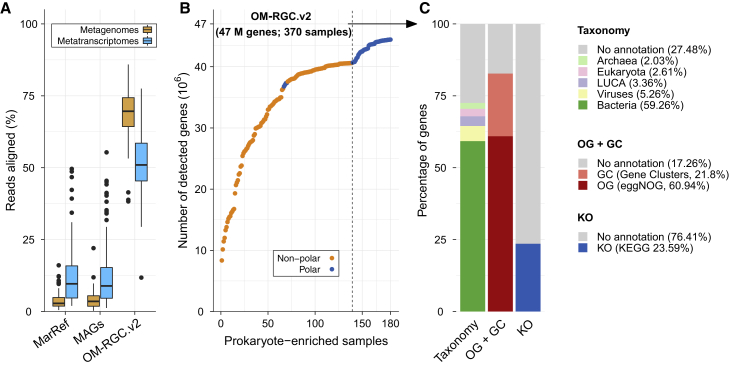
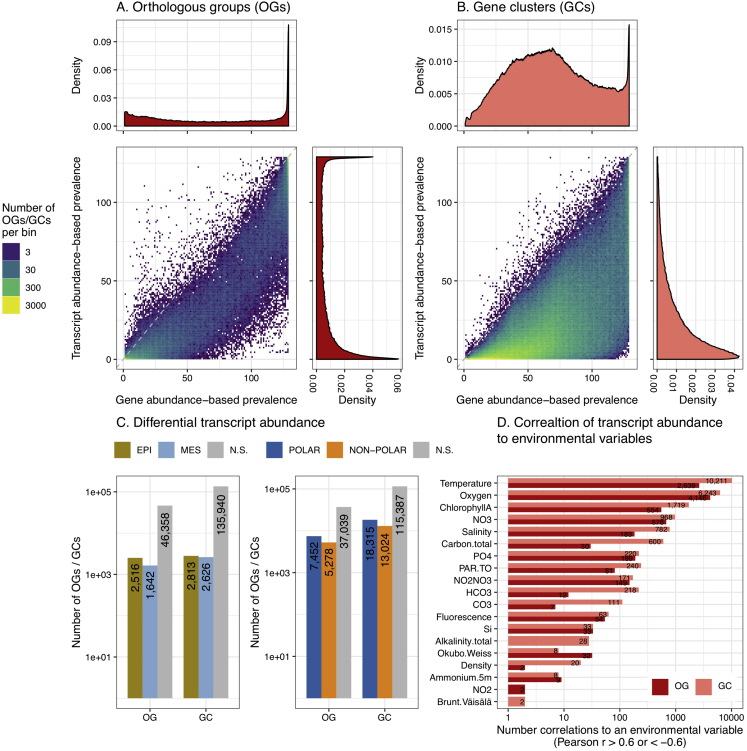
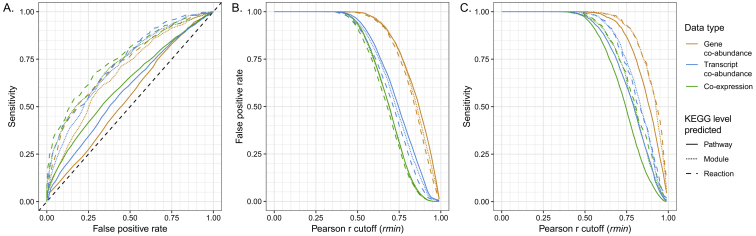
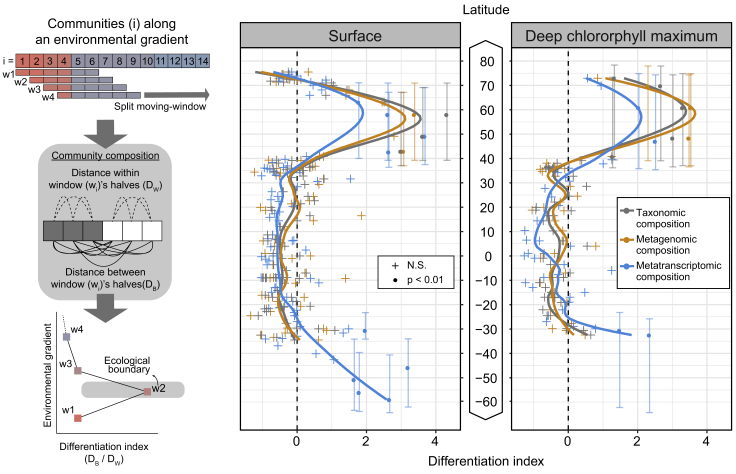
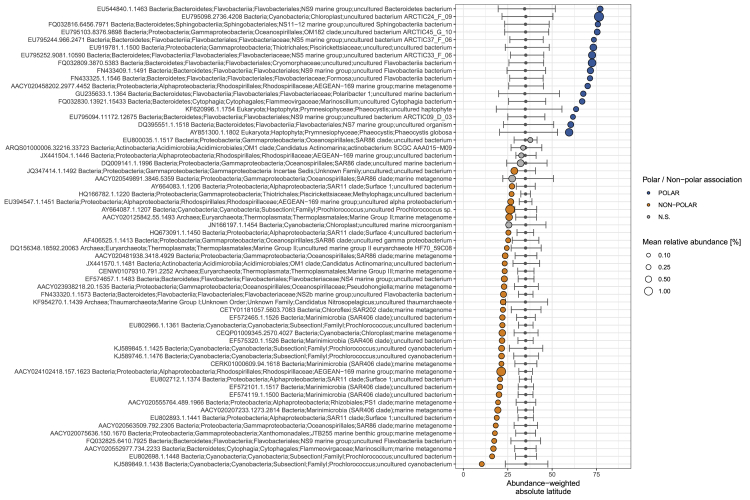
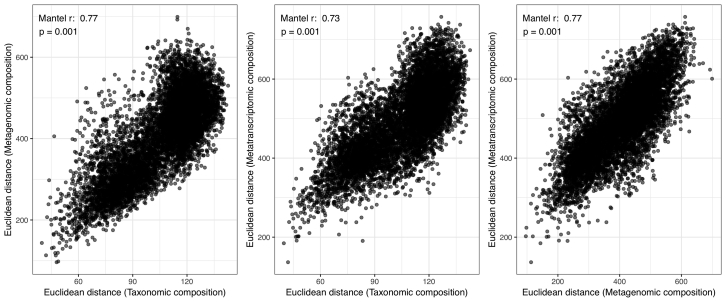
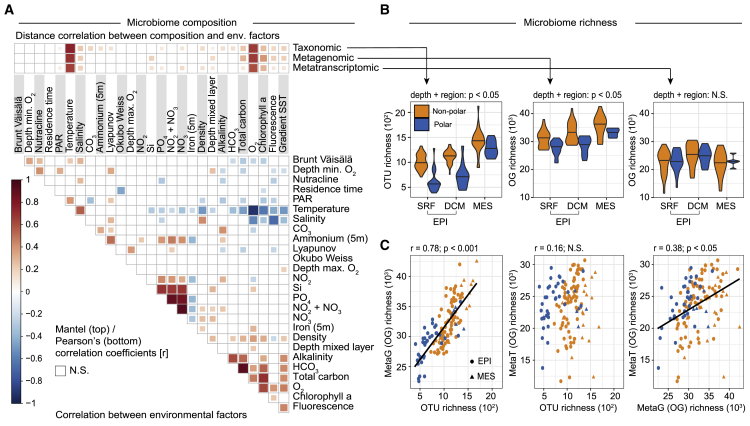
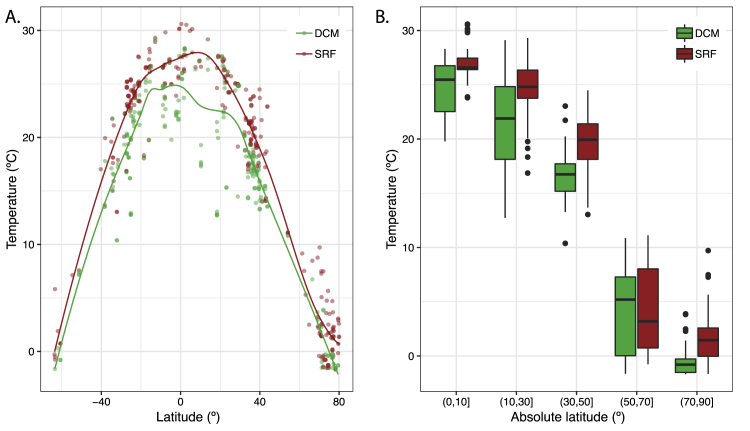
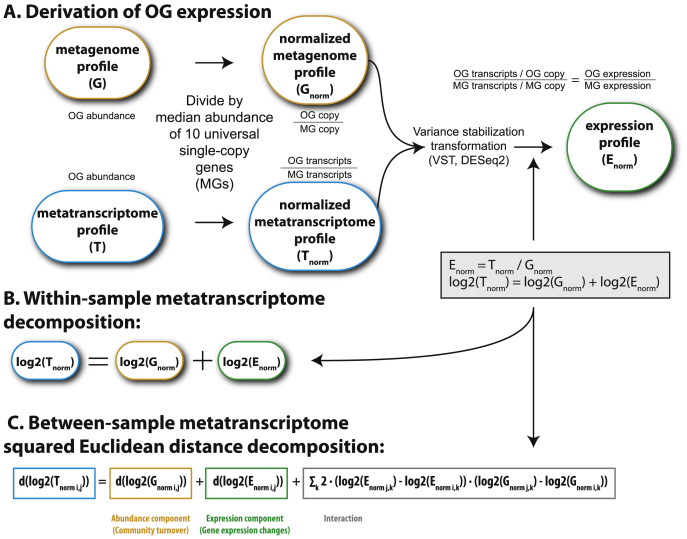
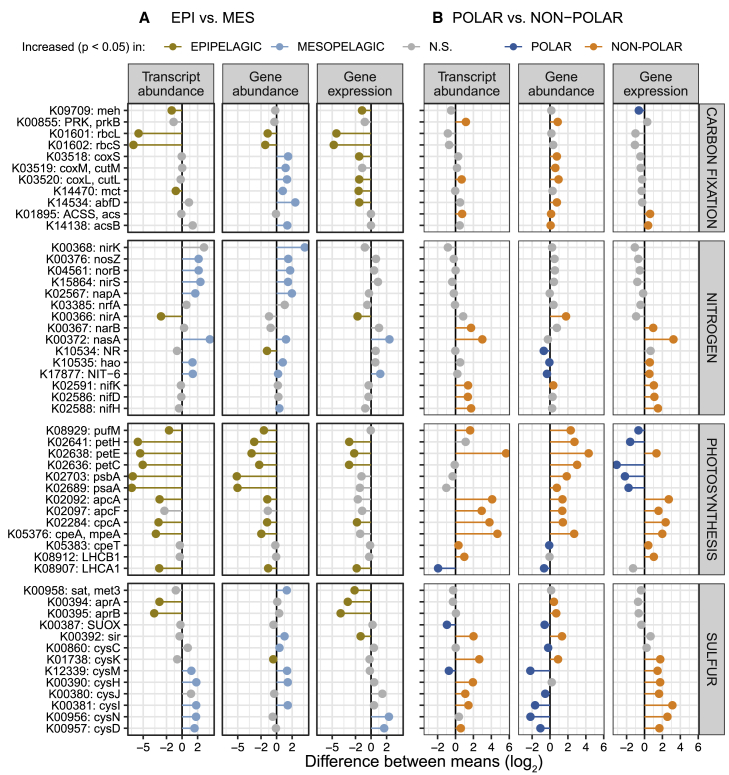
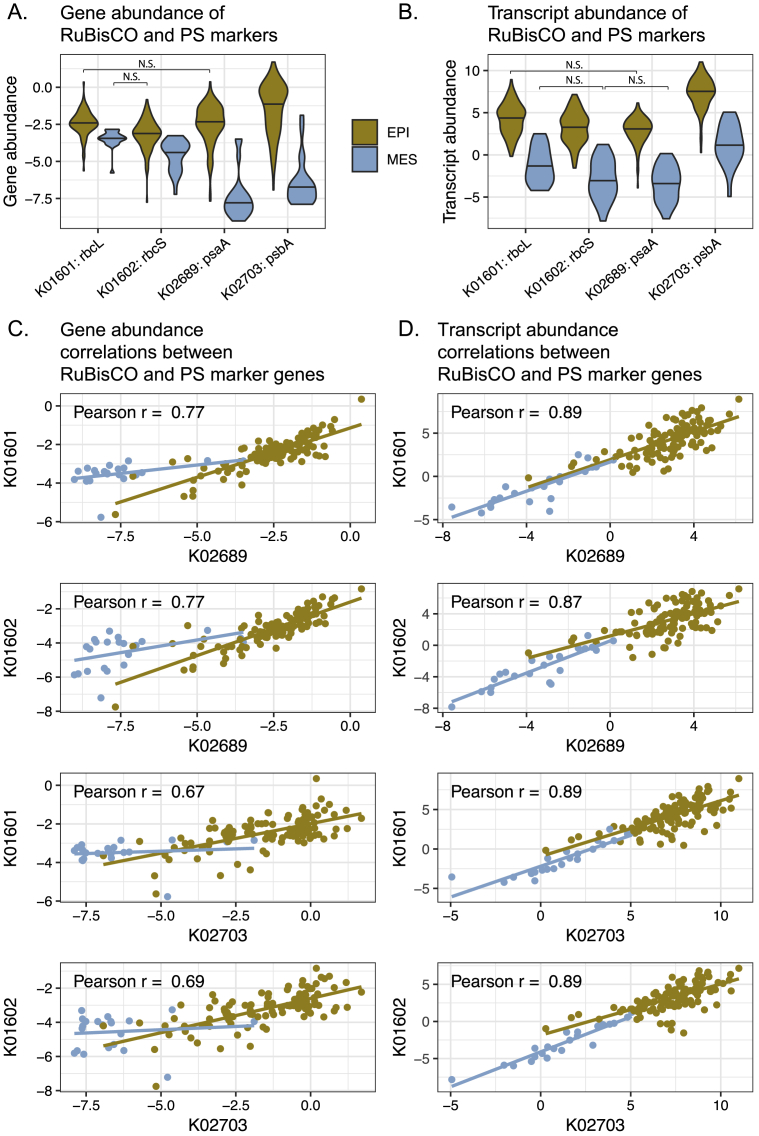
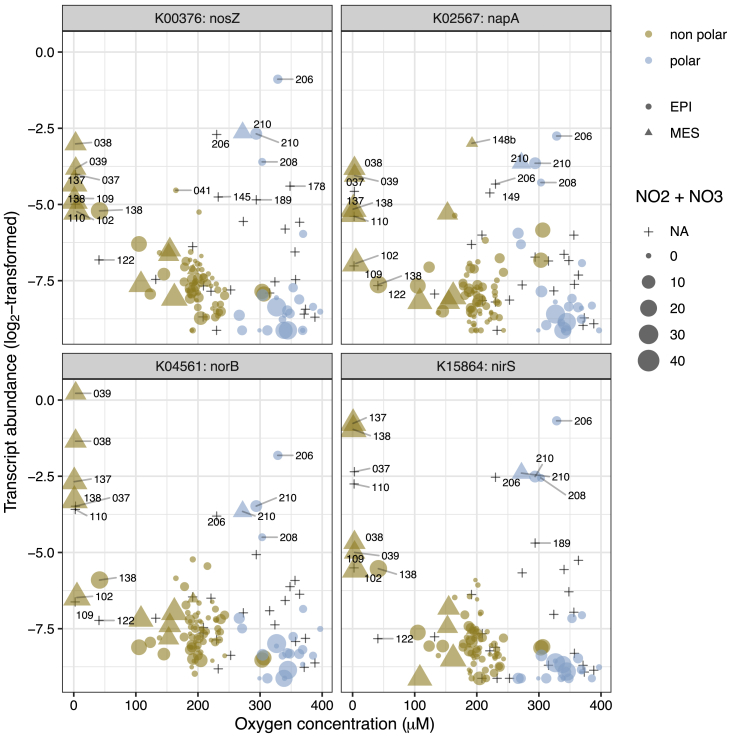
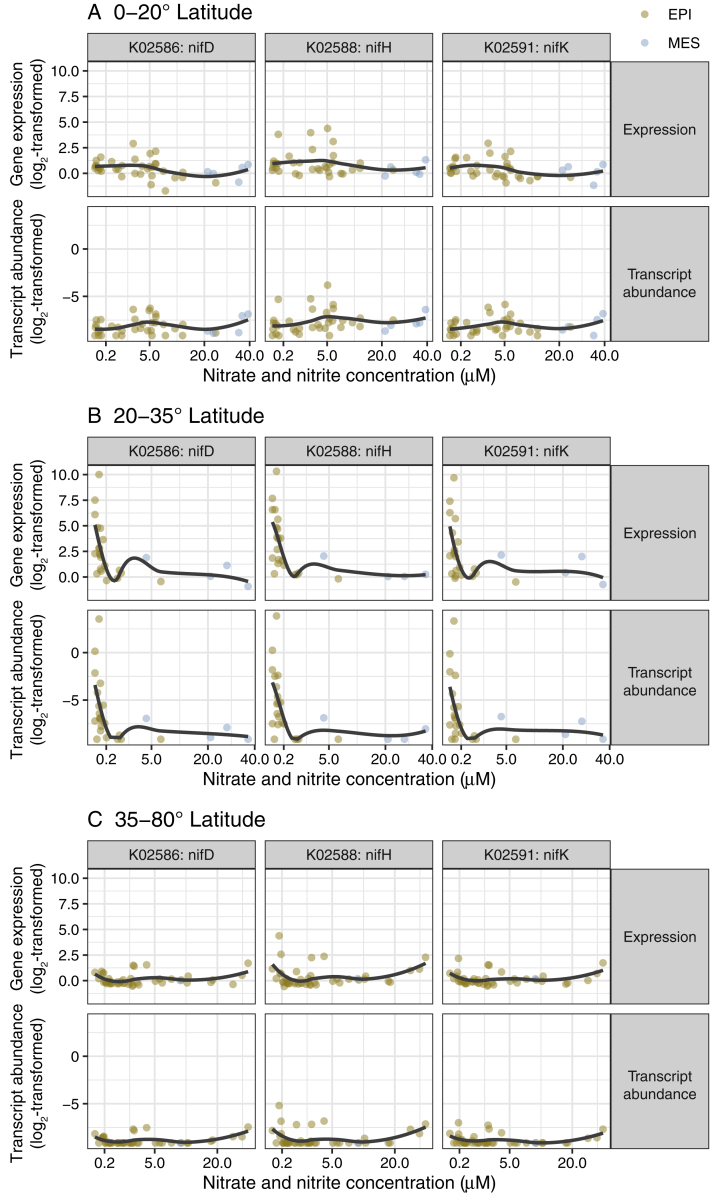
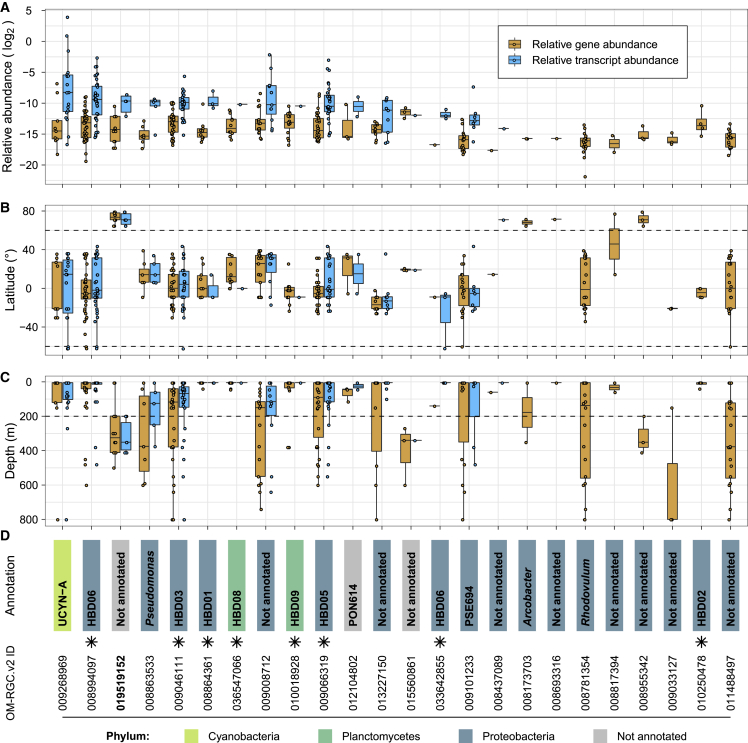
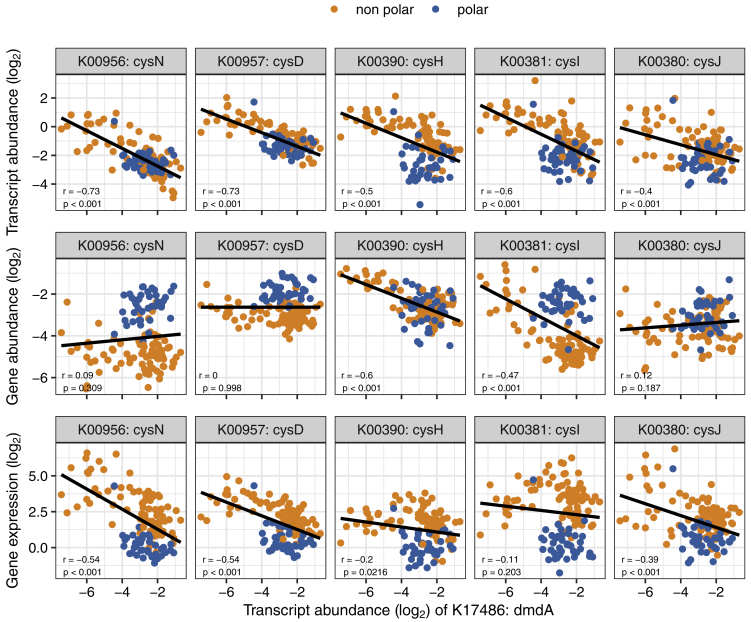
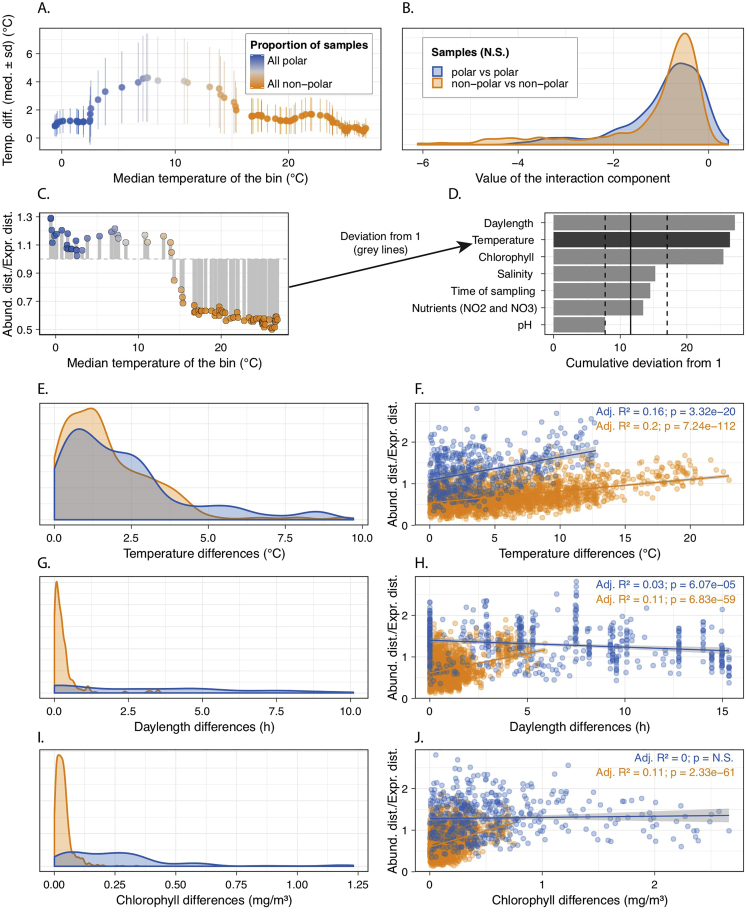
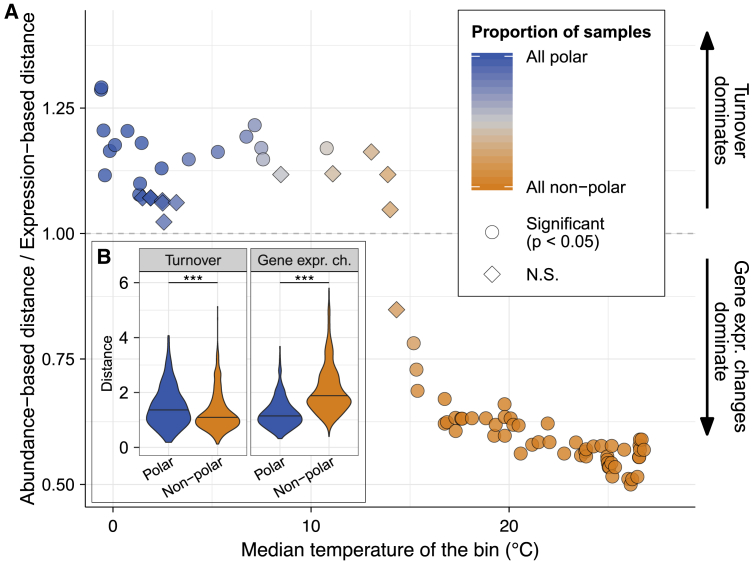
Ocean microbial communities strongly influence the biogeochemistry, food webs, and climate of our planet. Despite recent advances in understanding their taxonomic and genomic compositions, little is known about how their transcriptomes vary globally. Here, we present a dataset of 187 metatranscriptomes and 370 metagenomes from 126 globally distributed sampling stations and establish a resource of 47 million genes to study community-level transcriptomes across depth layers from pole-to-pole. We examine gene expression changes and community turnover as the underlying mechanisms shaping community transcriptomes along these axes of environmental variation and show how their individual contributions differ for multiple biogeochemically relevant processes. Furthermore, we find the relative contribution of gene expression changes to be significantly lower in polar than in non-polar waters and hypothesize that in polar regions, alterations in community activity in response to ocean warming will be driven more strongly by changes in organismal composition than by gene regulatory mechanisms. VIDEO ABSTRACT.
Keywords: Tara Oceans; biogeochemistry; community turnover; eco-systems biology; gene expression change; global ocean microbiome; metagenome; metatranscriptome; microbial ecology; ocean warming.
Copyright © 2019 The Author(s). Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Alberti A., Poulain J., Engelen S., Labadie K., Romac S., Ferrera I., Albini G., Aury J.-M., Belser C., Bertrand A., Genoscope Technical Team. Tara Oceans Consortium Coordinators Viral to metazoan marine plankton nucleotide sequences from the Tara Oceans expedition. Sci. Data. 2017;4:170093. - PMC - PubMed
-
- Alexander M.A., Scott J.D., Friedland K.D., Mills K.E., Nye J.A., Pershing A.J., Thomas A.C. Projected sea surface temperatures over the 21st century: Changes in the mean, variability and extremes for large marine ecosystem regions of Northern Oceans. Elem. Sci. Anth. 2018;6:9.
-
- Anderson R., Mawji E., Cutter G., Measures C., Jeandel C. GEOTRACES: Changing the Way We Explore Ocean Chemistry. Oceanography. 2014;27:50–61.
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
