

Clonal Decomposition and DNA Replication States Defined by Scaled Single-Cell Genome Sequencing
- PMID: 31730858
- PMCID: PMC6912164
- DOI: 10.1016/j.cell.2019.10.026
Clonal Decomposition and DNA Replication States Defined by Scaled Single-Cell Genome Sequencing
Abstract
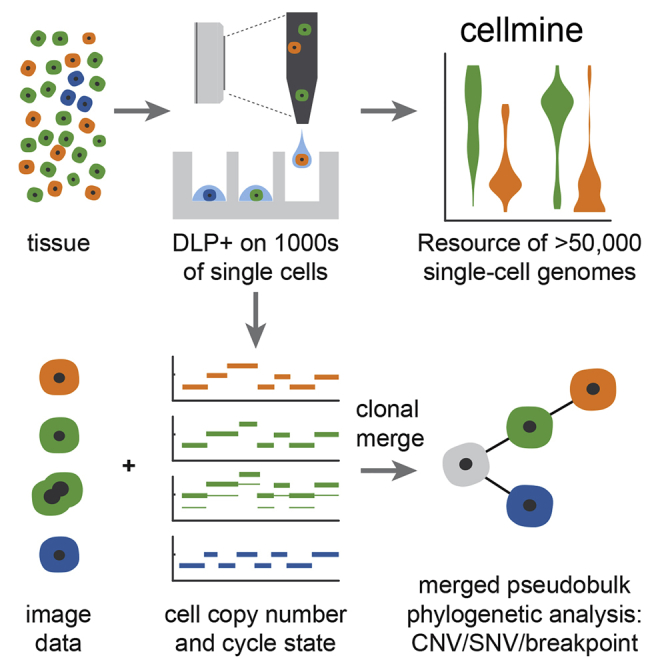
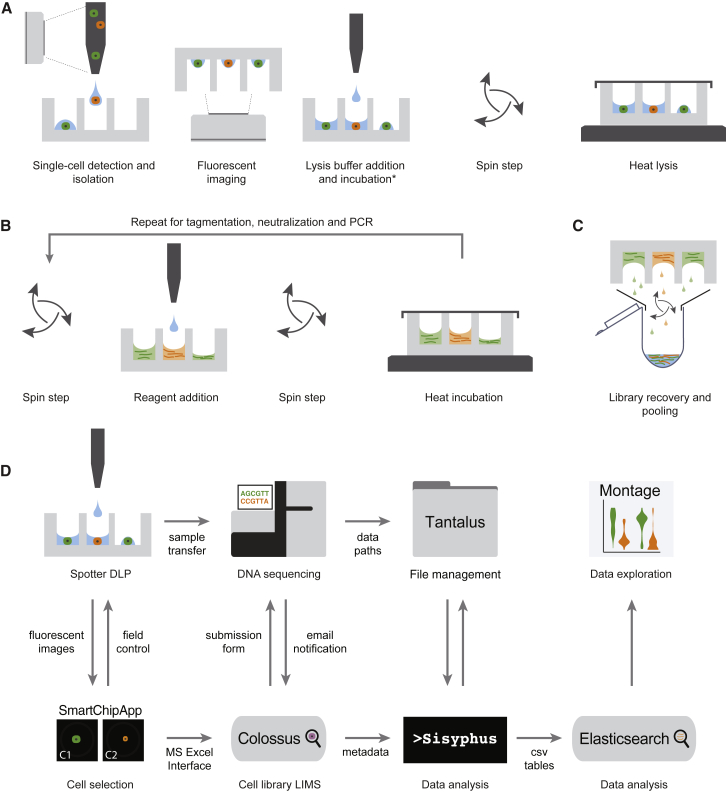
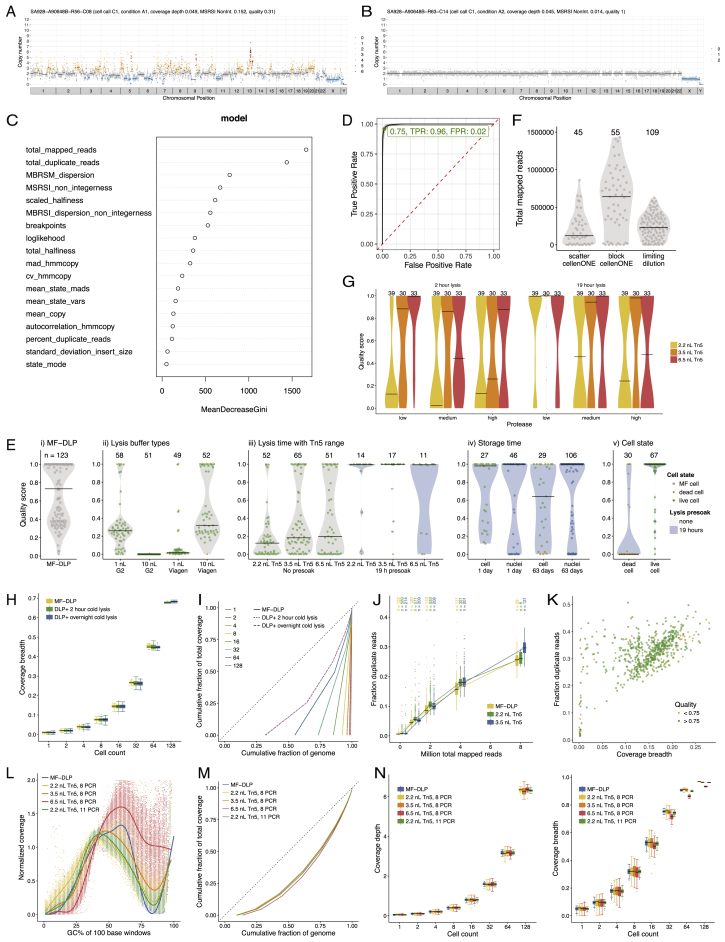
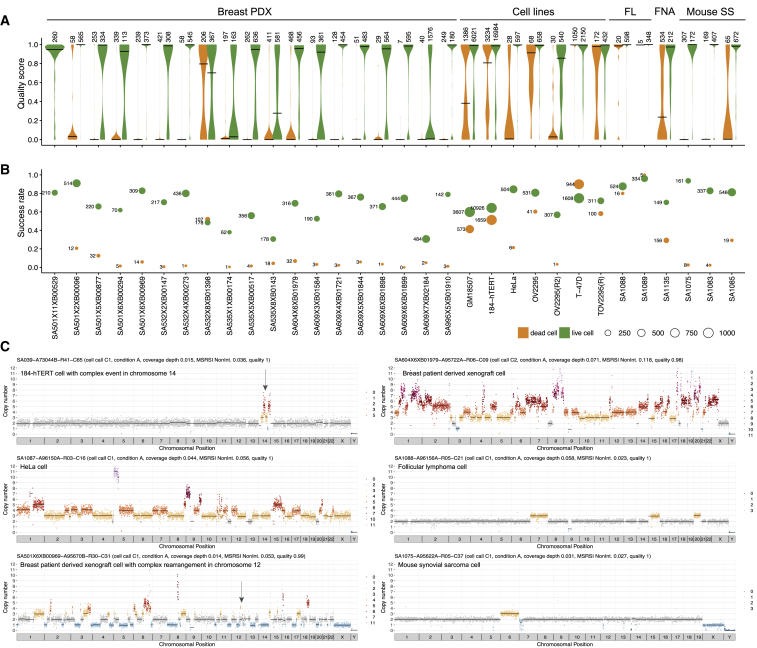
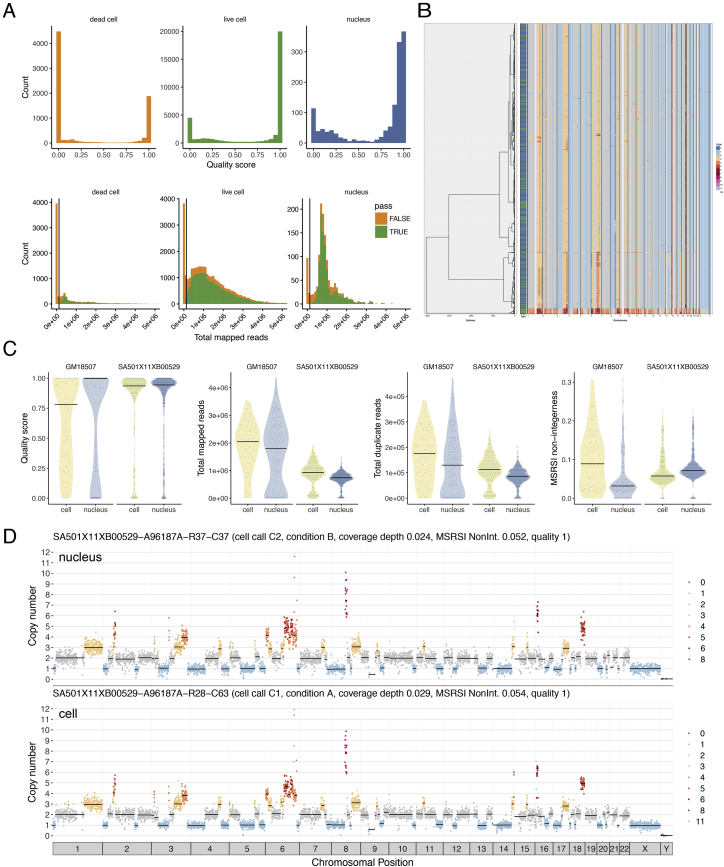
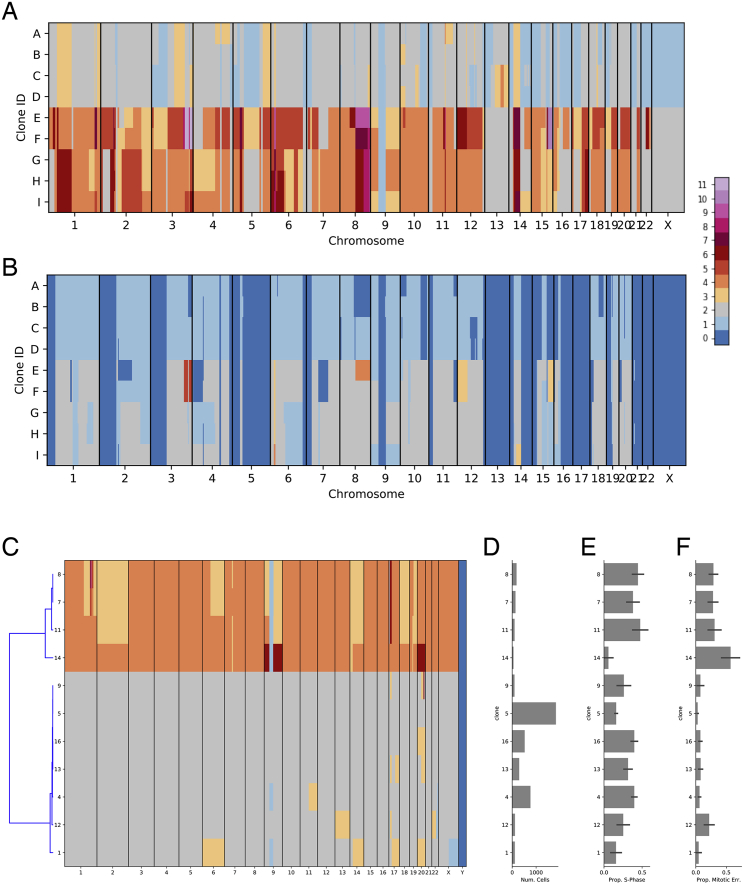
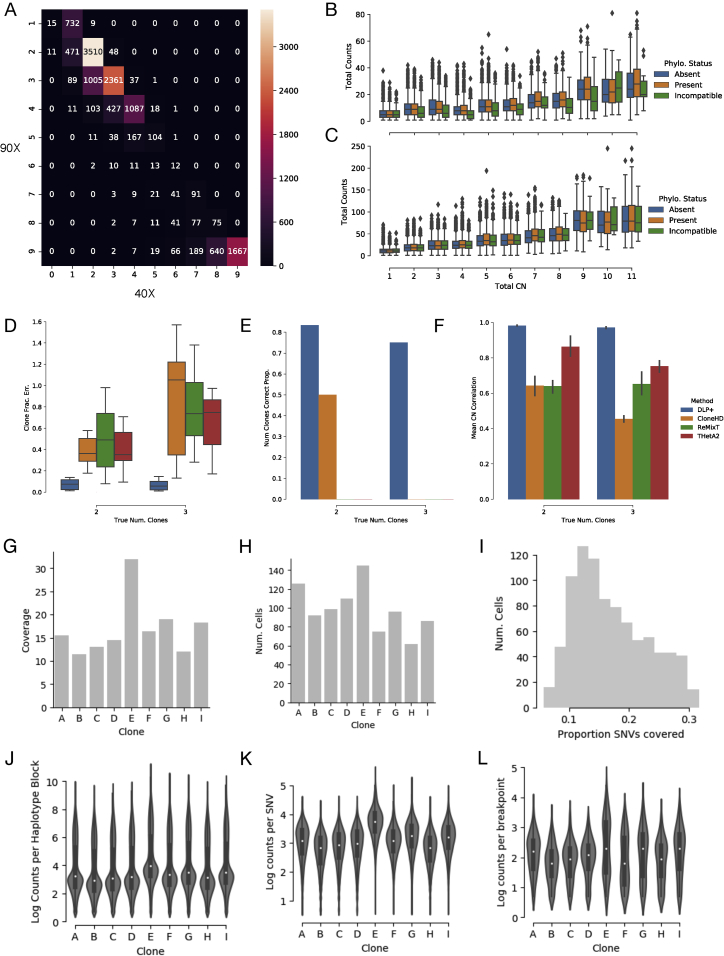
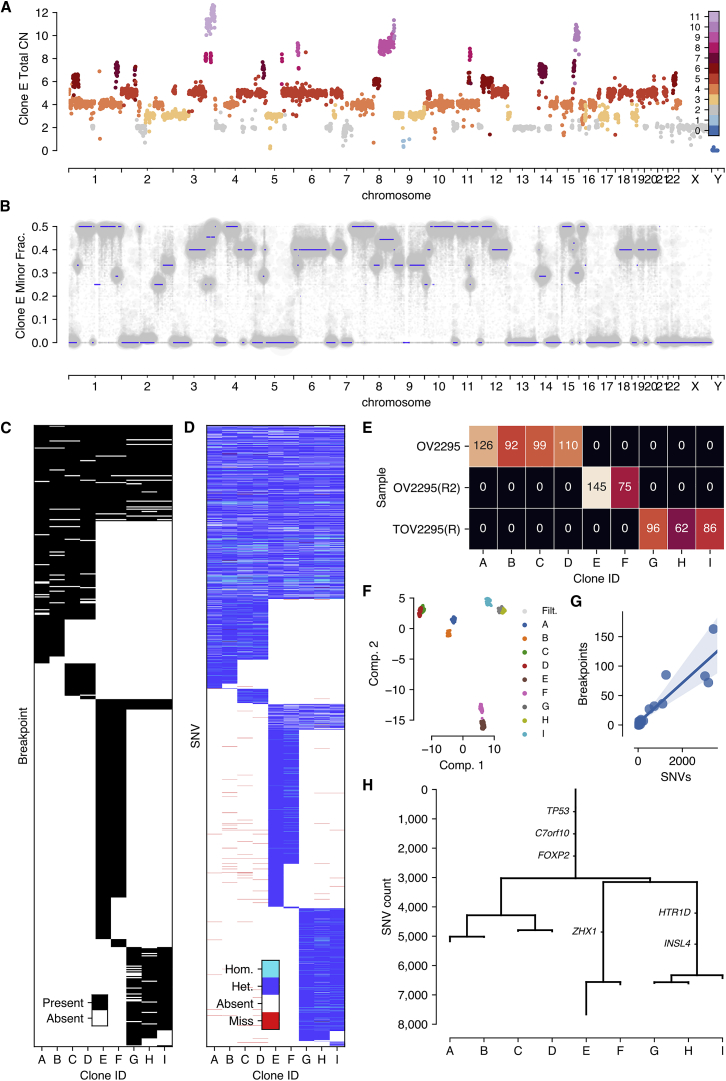
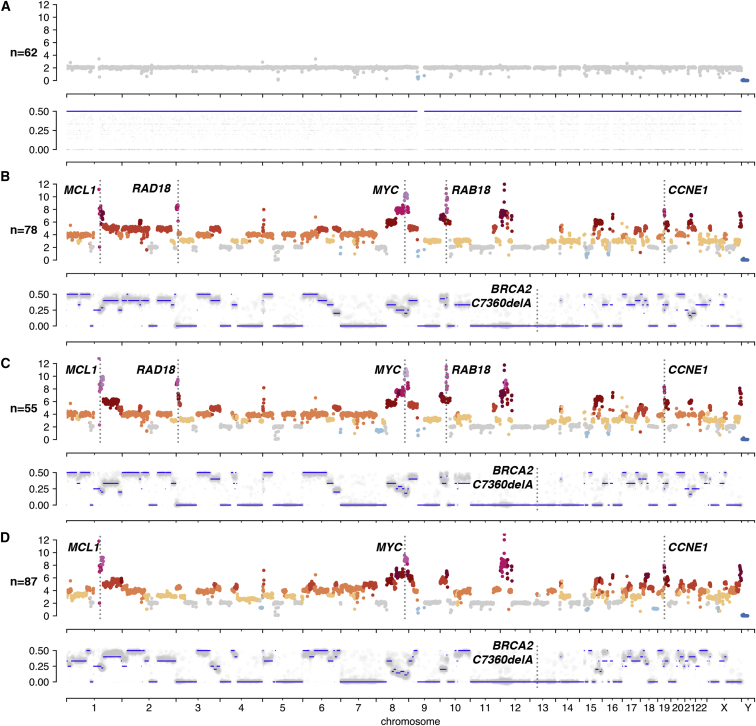
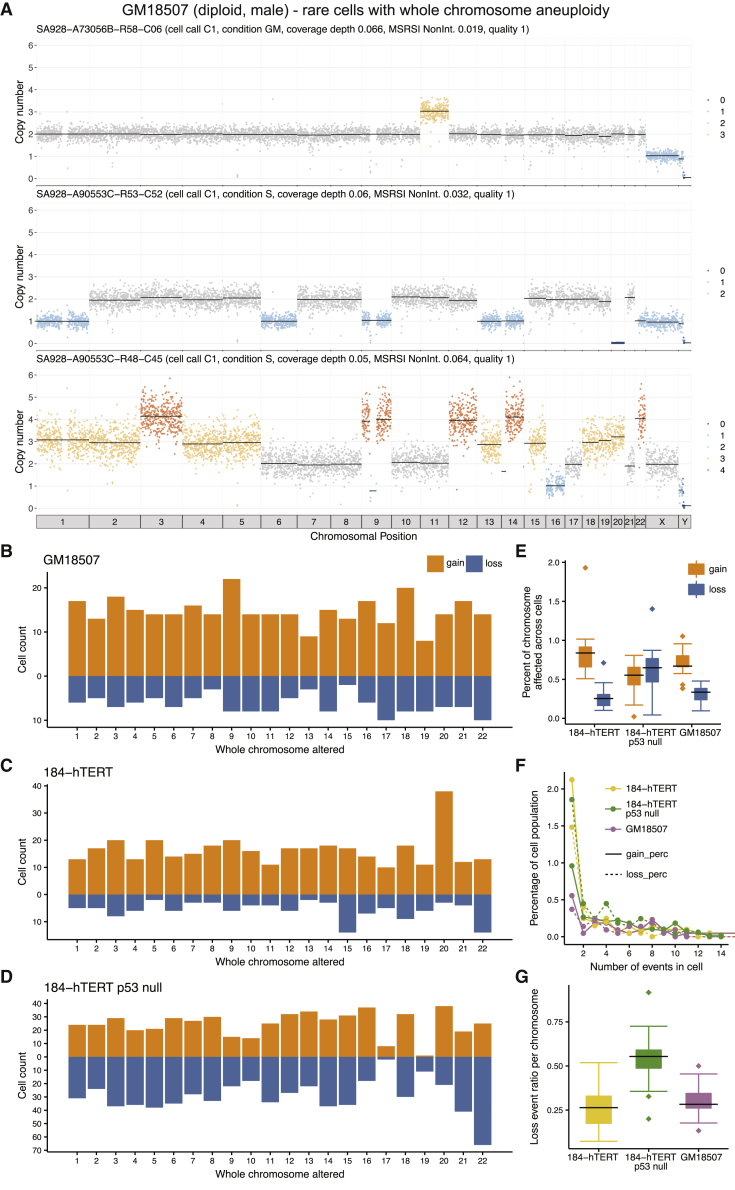
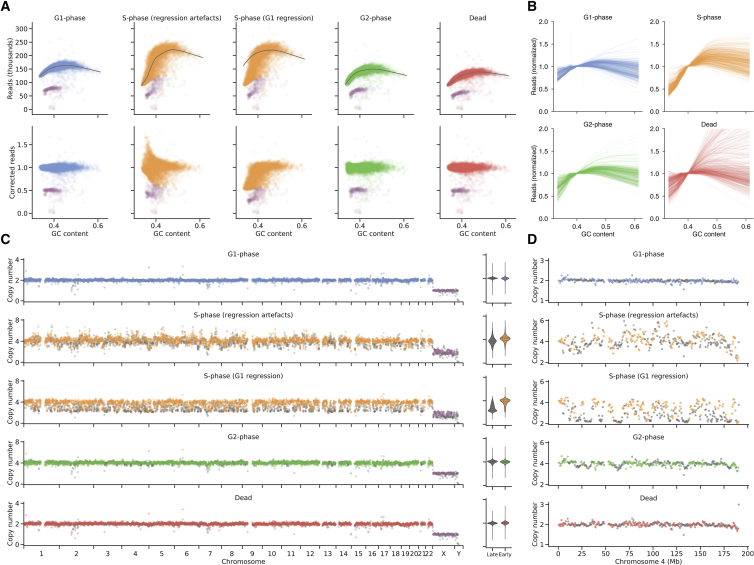
Accurate measurement of clonal genotypes, mutational processes, and replication states from individual tumor-cell genomes will facilitate improved understanding of tumor evolution. We have developed DLP+, a scalable single-cell whole-genome sequencing platform implemented using commodity instruments, image-based object recognition, and open source computational methods. Using DLP+, we have generated a resource of 51,926 single-cell genomes and matched cell images from diverse cell types including cell lines, xenografts, and diagnostic samples with limited material. From this resource we have defined variation in mitotic mis-segregation rates across tissue types and genotypes. Analysis of matched genomic and image measurements revealed correlations between cellular morphology and genome ploidy states. Aggregation of cells sharing copy number profiles allowed for calculation of single-nucleotide resolution clonal genotypes and inference of clonal phylogenies and avoided the limitations of bulk deconvolution. Finally, joint analysis over the above features defined clone-specific chromosomal aneuploidy in polyclonal populations.
Keywords: DNA sequencing; aneuploidy; cancer genomics; cell cycle; copy number; genomic instability; single cell; tumor evolution; tumor heterogeneity.
Copyright © 2019 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
S.P.S. and S.A. are founders and shareholders of Contextual Genomics Inc.
Figures

Comment in
-
Amplification-free single-cell whole-genome sequencing gets a makeover.Nat Methods. 2020 Jan;17(1):27. doi: 10.1038/s41592-019-0722-2. Nat Methods. 2020. PMID: 31907478 No abstract available.
References
-
- Ackerman M., Ben-David S. Which data sets are clusterable?: A theoretical study of clusterability. Journal of Machine Learning Research. 2009;5:1–8.
-
- Breiman L. Random Forests. Mach. Learn. 2001;45:5–32.
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
