

Distinct effects of ruxolitinib and interferon-alpha on murine JAK2V617F myeloproliferative neoplasm hematopoietic stem cell populations
- PMID: 31732720
- PMCID: PMC9221569
- DOI: 10.1038/s41375-019-0638-y
Distinct effects of ruxolitinib and interferon-alpha on murine JAK2V617F myeloproliferative neoplasm hematopoietic stem cell populations
Abstract
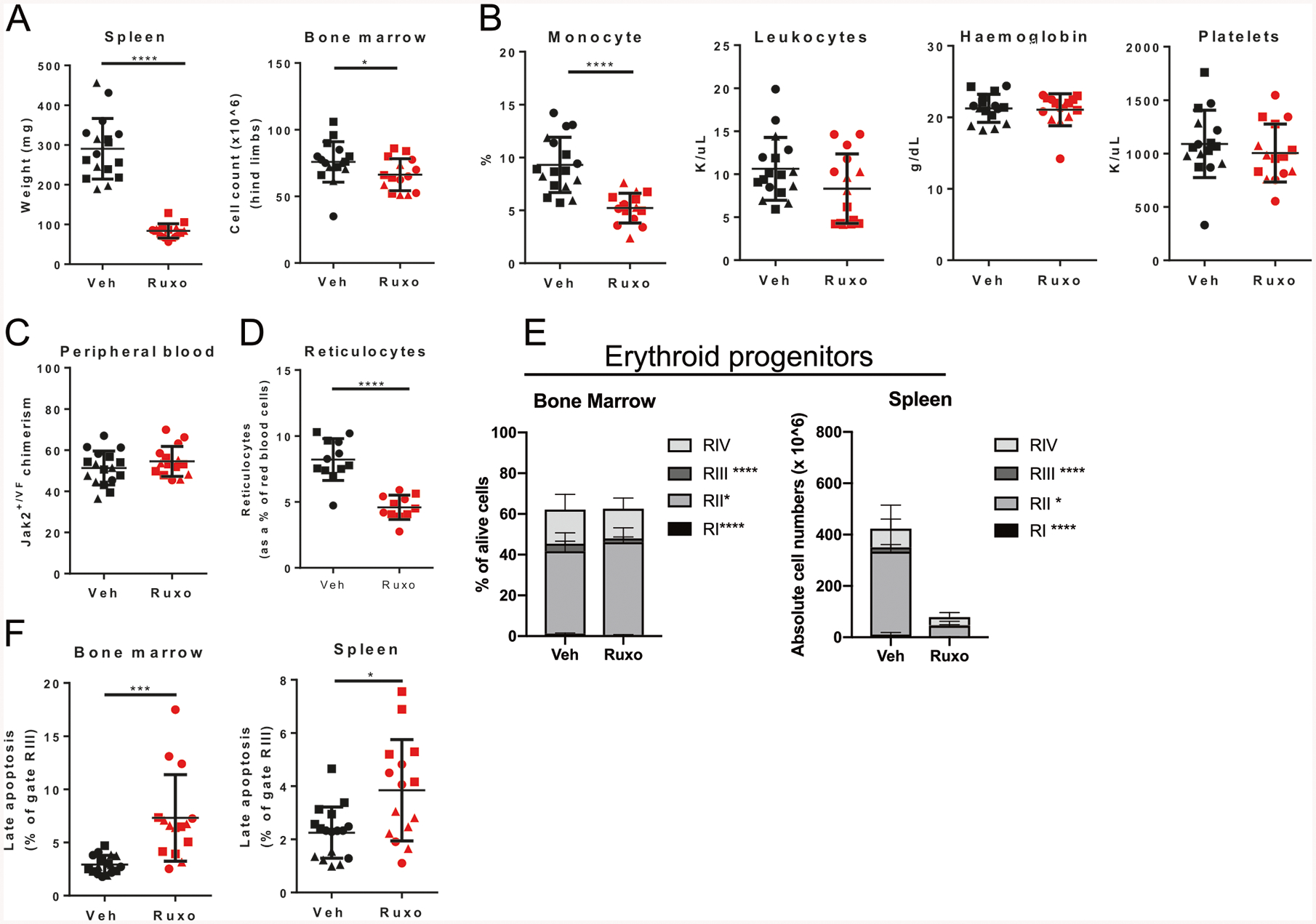
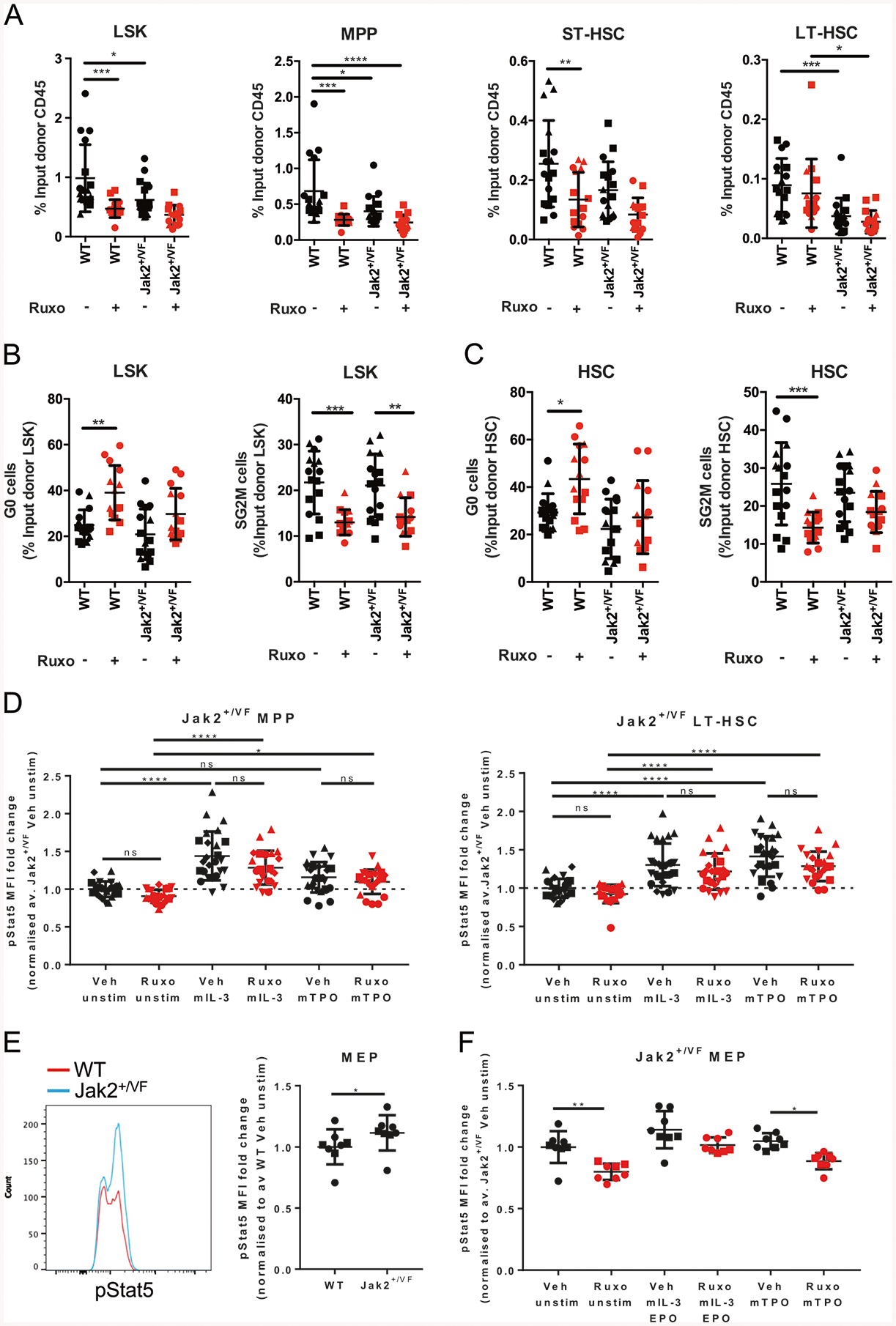
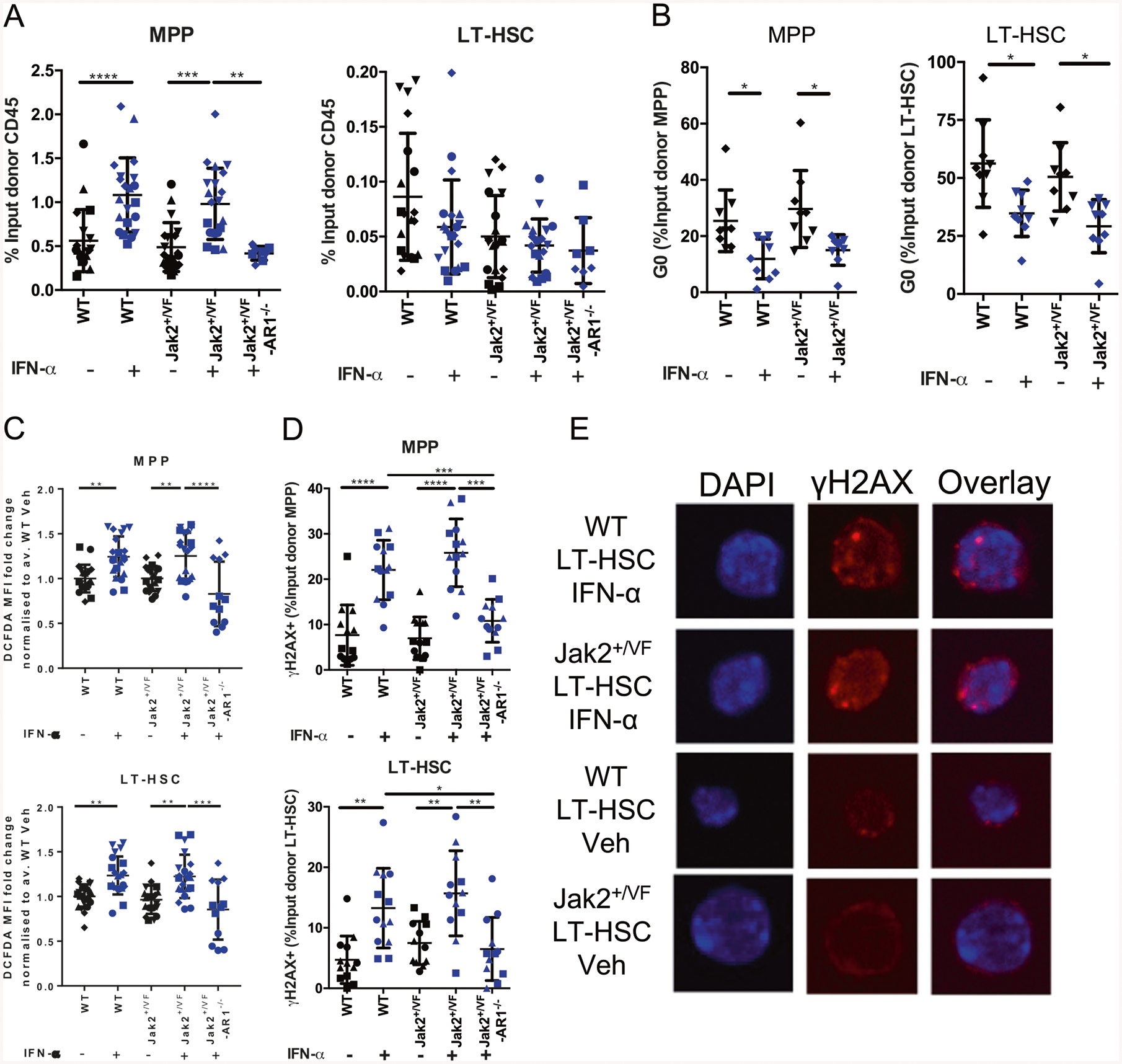
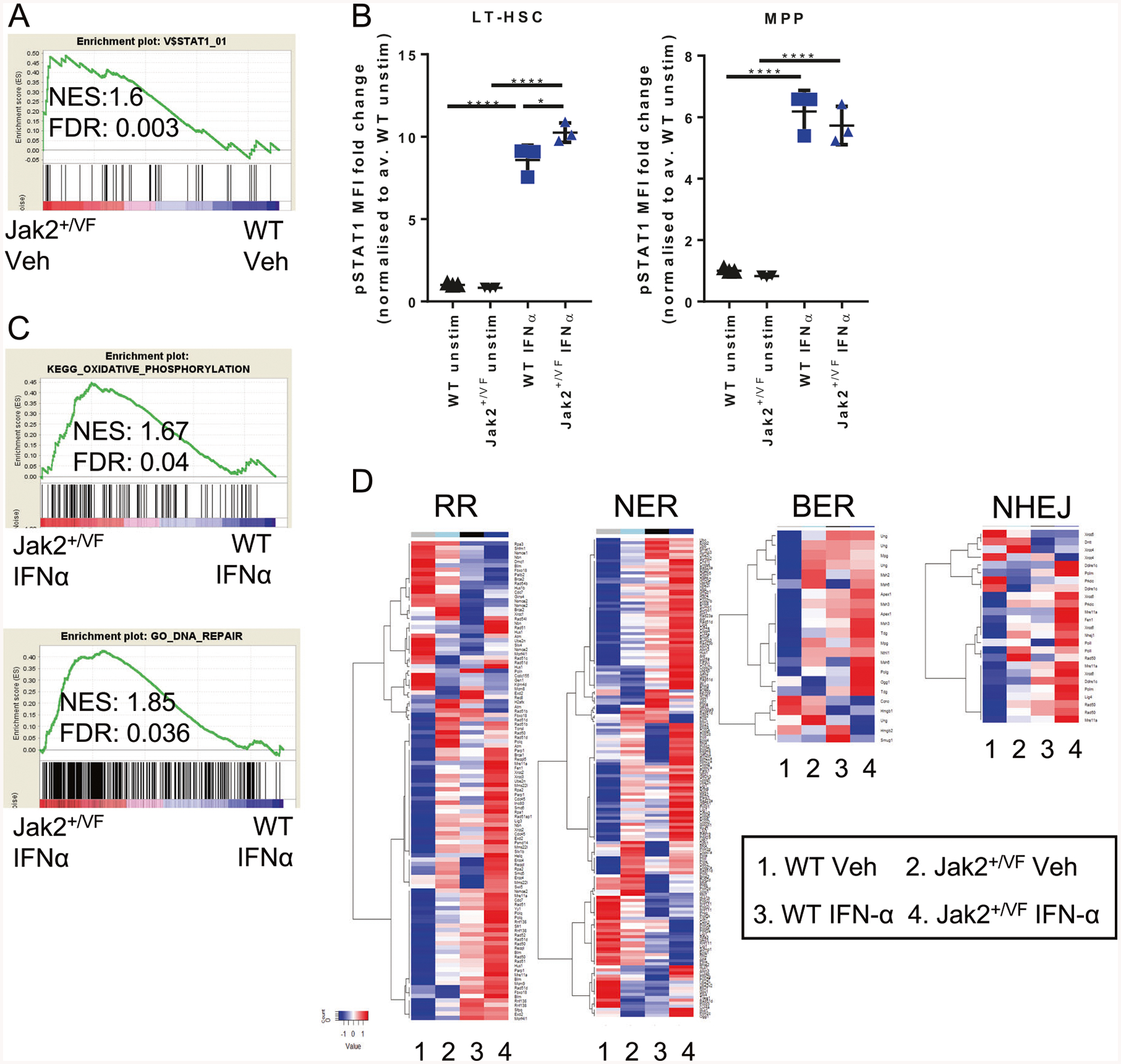
JAK2V617F is the most common mutation in patients with BCR-ABL negative myeloproliferative neoplasms (MPNs). The eradication of JAK2V617F hematopoietic stem cells (HSCs) is critical for achieving molecular remissions and cure. We investigate the distinct effects of two therapies, ruxolitinib (JAK1/2 inhibitor) and interferon-alpha (IFN-α), on the disease-initiating HSC population. Whereas ruxolitinib inhibits Stat5 activation in erythroid progenitor populations, it fails to inhibit this same pathway in HSCs. In contrast, IFN-α has direct effects on HSCs. Furthermore, STAT1 phosphorylation and pathway activation is greater after IFN-α stimulation in Jak2V617F murine HSCs with increased induction of reactive oxygen species, DNA damage and reduction in quiescence after chronic IFN-α treatment. Interestingly, ruxolitinib does not block IFN-α induced reactive oxygen species and DNA damage in Jak2V617F murine HSCs in vivo. This work provides a mechanistic rationale informing how pegylated IFN-α reduces JAK2V617F allelic burden in the clinical setting and may inform future clinical efforts to combine ruxolitinib with pegylated IFN-α in patients with MPN.
Conflict of interest statement
Figures



References
-
- Ishii T, Bruno E, Hoffman R, Xu M. Involvement of various hematopoietic-cell lineages by the JAK2V617F mutation in polycythemia vera. Blood. 2006;108:3128–34. - PubMed
-
- Baxter EJ, Scott LM, Campbell PJ, East C, Fourouclas N, Swanton S, et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet. 2005;365: 1054–61. - PubMed
-
- James C, Ugo V, Le Couedic JP, Staerk J, Delhommeau F, Lacout C, et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature. 2005;434:1144–8. - PubMed
-
- Kralovics R, Passamonti F, Buser AS, Teo S-S, Tiedt R, Passweg JR, et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders. New Engl J Med. 2005;352:1779–90. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
