

Prion-Like Propagation of Protein Misfolding and Aggregation in Amyotrophic Lateral Sclerosis
- PMID: 31736708
- PMCID: PMC6838634
- DOI: 10.3389/fnmol.2019.00262
Prion-Like Propagation of Protein Misfolding and Aggregation in Amyotrophic Lateral Sclerosis
Erratum in
-
Corrigendum: Prion-Like Propagation of Protein Misfolding and Aggregation in Amyotrophic Lateral Sclerosis.Front Mol Neurosci. 2020 Jan 21;12:311. doi: 10.3389/fnmol.2019.00311. eCollection 2019. Front Mol Neurosci. 2020. PMID: 32038158 Free PMC article.
Abstract
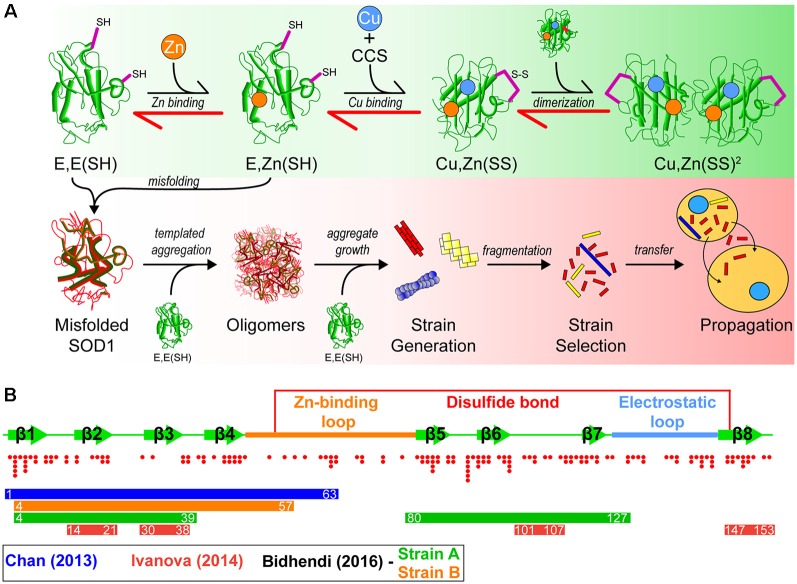
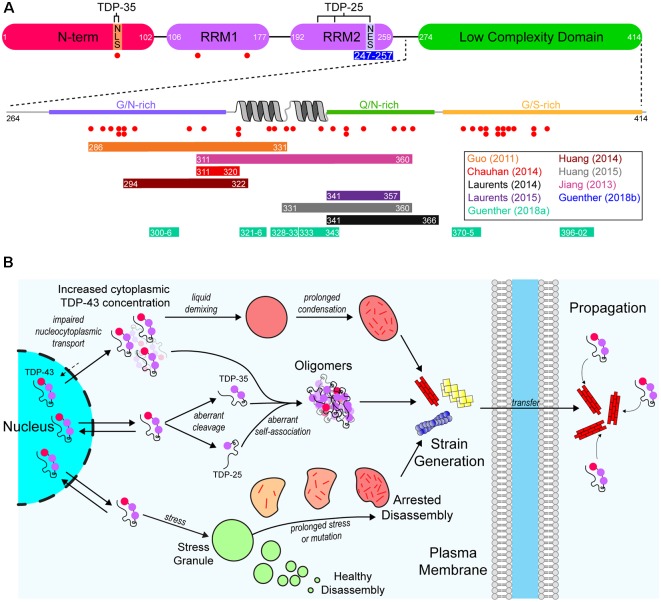
The discovery that prion protein can misfold into a pathological conformation that encodes structural information capable of both propagation and inducing severe neuropathology has revolutionized our understanding of neurodegenerative disease. Many neurodegenerative diseases with a protein misfolding component are now classified as "prion-like" owing to the propagation of both symptoms and protein aggregation pathology in affected individuals. The neuromuscular disorder amyotrophic lateral sclerosis (ALS) is characterized by protein inclusions formed by either TAR DNA-binding protein of 43 kDa (TDP-43), Cu/Zn superoxide dismutase (SOD1), or fused in sarcoma (FUS), in both upper and lower motor neurons. Evidence from in vitro, cell culture, and in vivo studies has provided strong evidence to support the involvement of a prion-like mechanism in ALS. In this article, we review the evidence suggesting that prion-like propagation of protein aggregation is a primary pathomechanism in ALS, focusing on the key proteins and genes involved in disease (TDP-43, SOD1, FUS, and C9orf72). In each case, we discuss the evidence ranging from biophysical studies to in vivo examinations of prion-like spreading. We suggest that the idiopathic nature of ALS may stem from its prion-like nature and that elucidation of the specific propagating protein assemblies is paramount to developing effective therapies.
Keywords: amyotrophic lateral sclerosis; prion; protein aggregation; protein misfolding; proteostasis.
Copyright © 2019 McAlary, Plotkin, Yerbury and Cashman.
Figures

References
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous