

What Do Neighbors Tell About You: The Local Context of Cis-Regulatory Modules Complicates Prediction of Regulatory Variants
- PMID: 31737053
- PMCID: PMC6834773
- DOI: 10.3389/fgene.2019.01078
What Do Neighbors Tell About You: The Local Context of Cis-Regulatory Modules Complicates Prediction of Regulatory Variants
Abstract
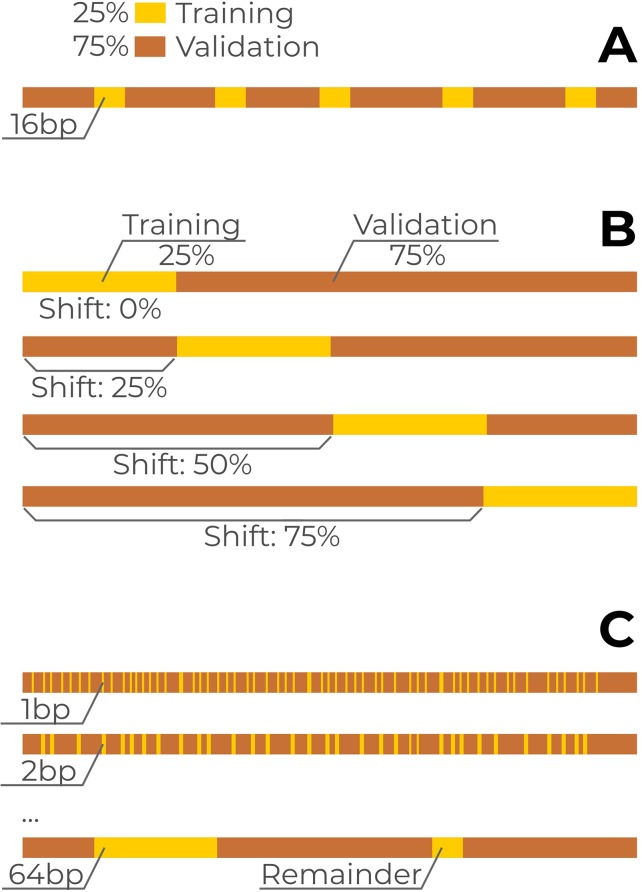
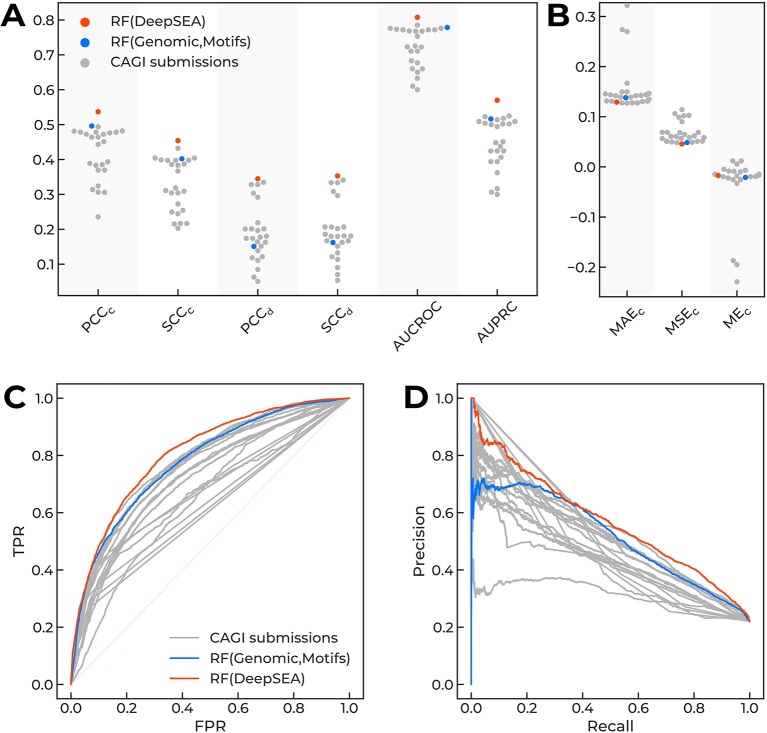
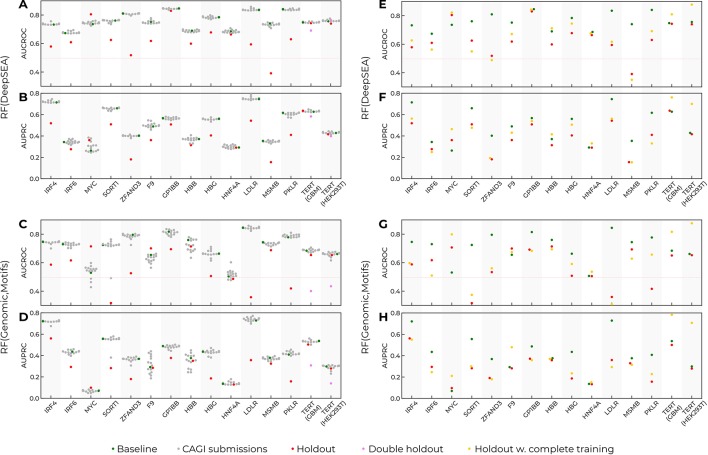
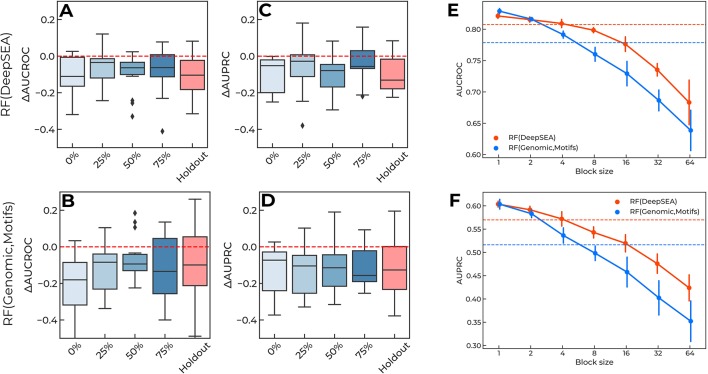
Many problems of modern genetics and functional genomics require the assessment of functional effects of sequence variants, including gene expression changes. Machine learning is considered to be a promising approach for solving this task, but its practical applications remain a challenge due to the insufficient volume and diversity of training data. A promising source of valuable data is a saturation mutagenesis massively parallel reporter assay, which quantitatively measures changes in transcription activity caused by sequence variants. Here, we explore the computational predictions of the effects of individual single-nucleotide variants on gene transcription measured in the massively parallel reporter assays, based on the data from the recent "Regulation Saturation" Critical Assessment of Genome Interpretation challenge. We show that the estimated prediction quality strongly depends on the structure of the training and validation data. Particularly, training on the sequence segments located next to the validation data results in the "information leakage" caused by the local context. This information leakage allows reproducing the prediction quality of the best CAGI challenge submissions with a fairly simple machine learning approach, and even obtaining notably better-than-random predictions using irrelevant genomic regions. Validation scenarios preventing such information leakage dramatically reduce the measured prediction quality. The performance at independent regulatory regions entirely excluded from the training set appears to be much lower than needed for practical applications, and even the performance estimation will become reliable only in the future with richer data from multiple reporters. The source code and data are available at https://bitbucket.org/autosomeru_cagi2018/cagi2018_regsat and https://genomeinterpretation.org/content/expression-variants.
Keywords: enhancers; machine learning; promoters; rSNP; regulatory variants; saturation mutagenesis massively parallel reporter assay.
Copyright © 2019 Penzar, Zinkevich, Vorontsov, Sitnik, Favorov, Makeev and Kulakovskiy.
Figures
References
-
- Boulesteix A.-L., Janitza S., Kruppa J., König I. R. (2012). Overview of random forest methodology and practical guidance with emphasis on computational biology and bioinformatics: Random forests in bioinformatics. WIREs Data Mining Knowl. Discovery 2 (6), 493–507. 10.1002/widm.1072 - DOI
-
- Cawley G. C., Talbot N. L. (2010). On over-fitting in model selection and subsequent selection bias in performance evaluation. J. Mach. Learn. Res. 11, 2079–2107.
Grants and funding
LinkOut - more resources
Full Text Sources