

Antibiotic Resistance and Epigenetics: More to It than Meets the Eye
- PMID: 31740560
- PMCID: PMC6985748
- DOI: 10.1128/AAC.02225-19
Antibiotic Resistance and Epigenetics: More to It than Meets the Eye
Abstract
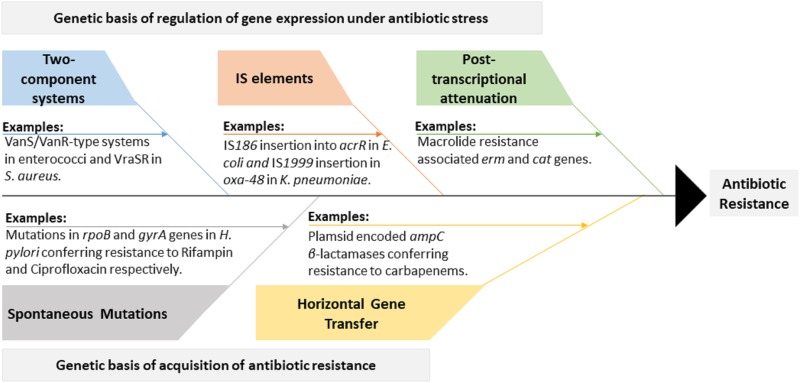
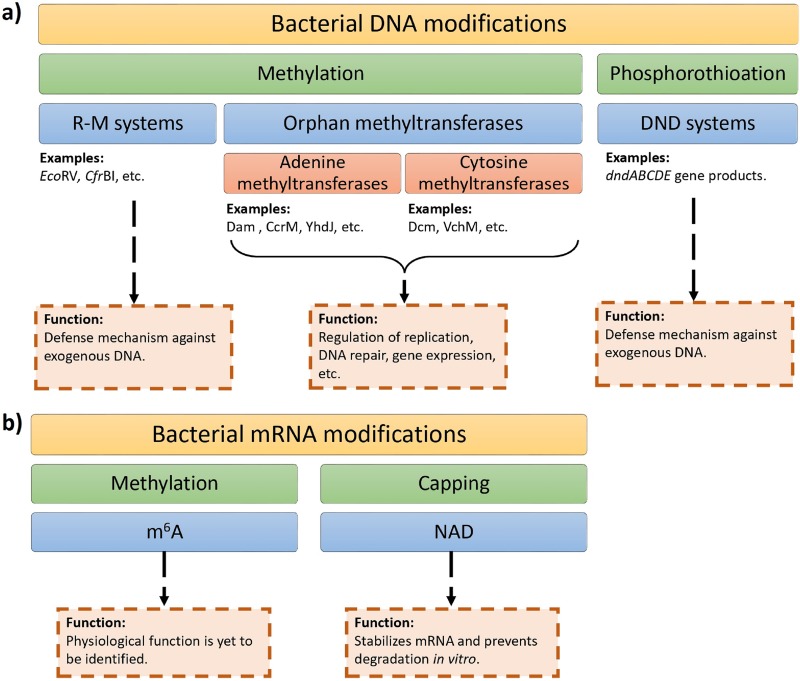
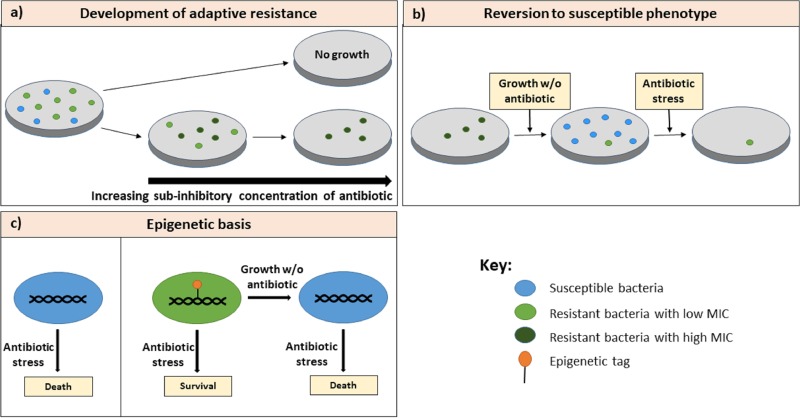
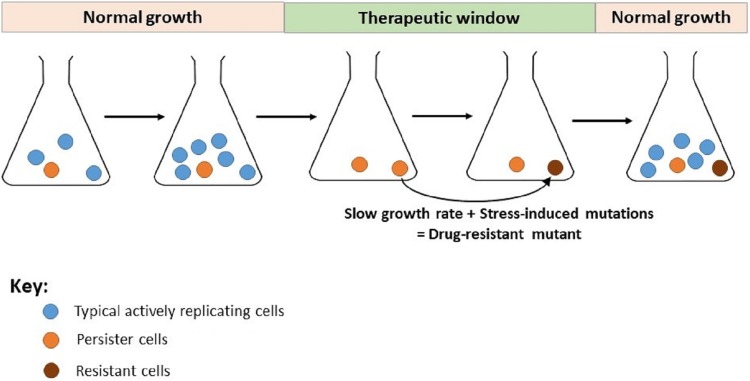
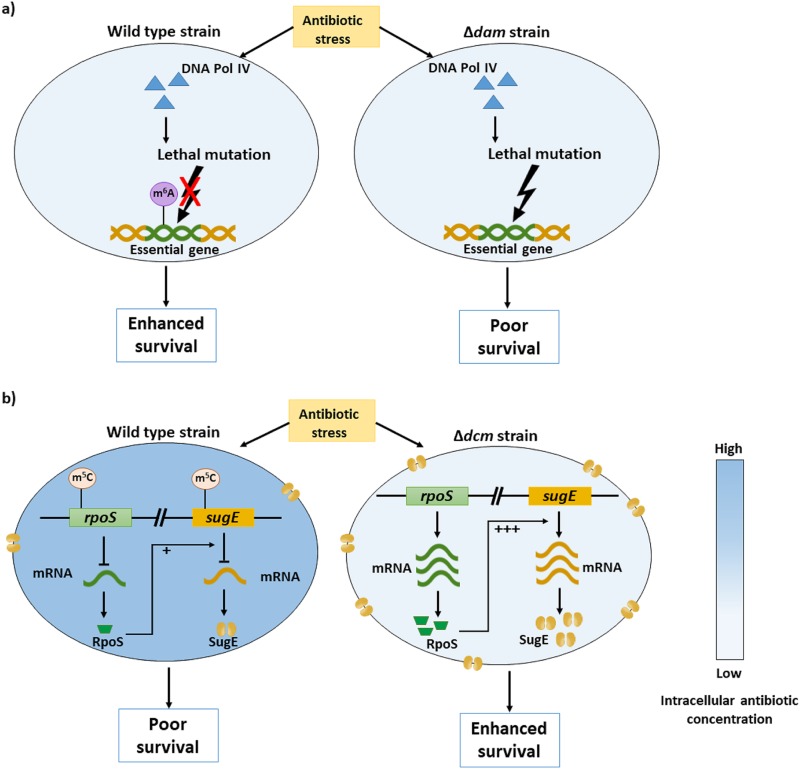
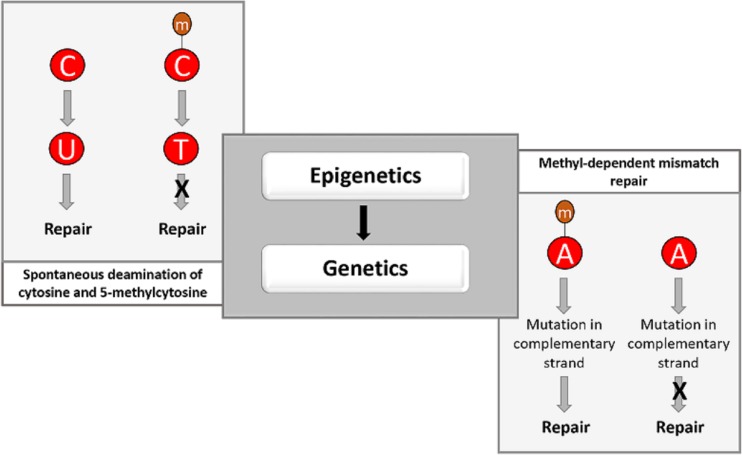
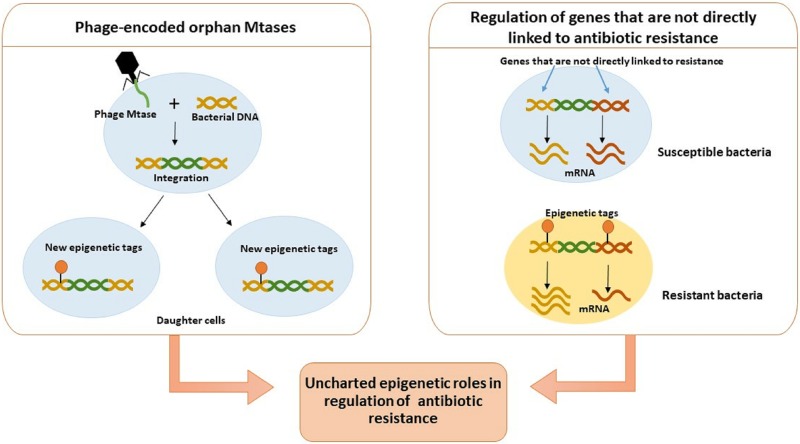
The discovery of antibiotics in the last century is considered one of the most important achievements in the history of medicine. Antibiotic usage has significantly reduced morbidity and mortality associated with bacterial infections. However, inappropriate use of antibiotics has led to emergence of antibiotic resistance at an alarming rate. Antibiotic resistance is regarded as a major health care challenge of this century. Despite extensive research, well-documented biochemical mechanisms and genetic changes fail to fully explain mechanisms underlying antibiotic resistance. Several recent reports suggest a key role for epigenetics in the development of antibiotic resistance in bacteria. The intrinsic heterogeneity as well as transient nature of epigenetic inheritance provides a plausible backdrop for high-paced emergence of drug resistance in bacteria. The methylation of adenines and cytosines can influence mutation rates in bacterial genomes, thus modulating antibiotic susceptibility. In this review, we discuss a plethora of recently discovered epigenetic mechanisms and their emerging roles in antibiotic resistance. We also highlight specific epigenetic mechanisms that merit further investigation for their role in antibiotic resistance.
Keywords: adaptive resistance; antibiotic resistance; antibiotics; bacterial epigenetics; methylation.
Copyright © 2020 American Society for Microbiology.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
