

Widespread long-range cis-regulatory elements in the maize genome
- PMID: 31740773
- PMCID: PMC6904520
- DOI: 10.1038/s41477-019-0547-0
Widespread long-range cis-regulatory elements in the maize genome
Erratum in
-
Author Correction: Widespread long-range cis-regulatory elements in the maize genome.Nat Plants. 2020 Mar;6(3):328. doi: 10.1038/s41477-020-0600-z. Nat Plants. 2020. PMID: 32029893
Abstract
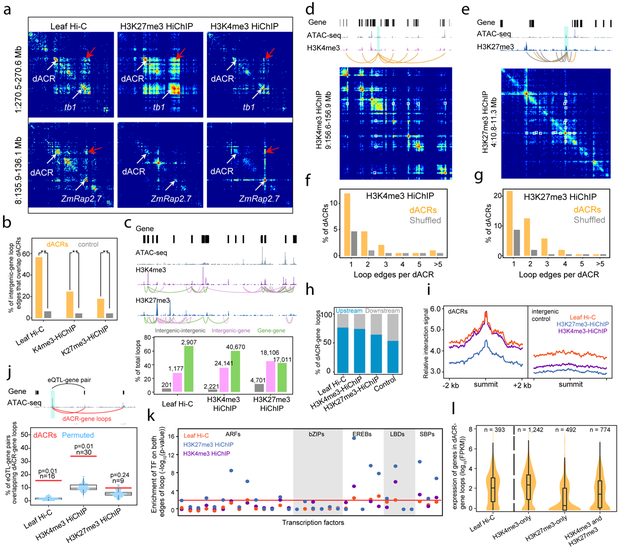
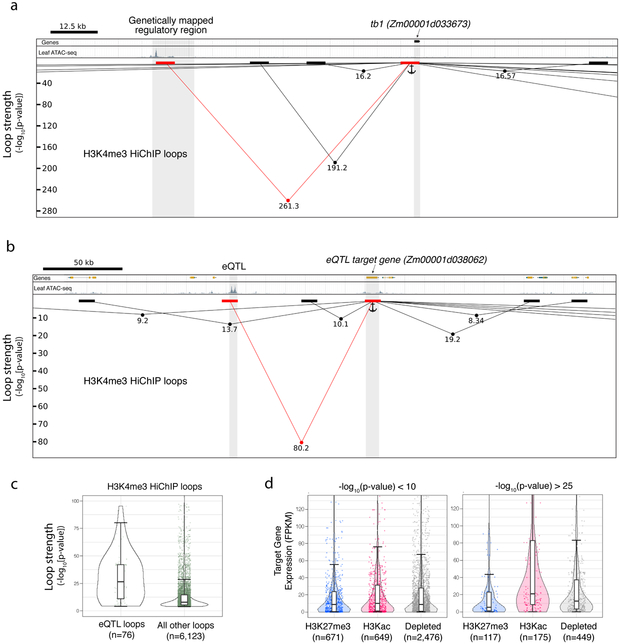
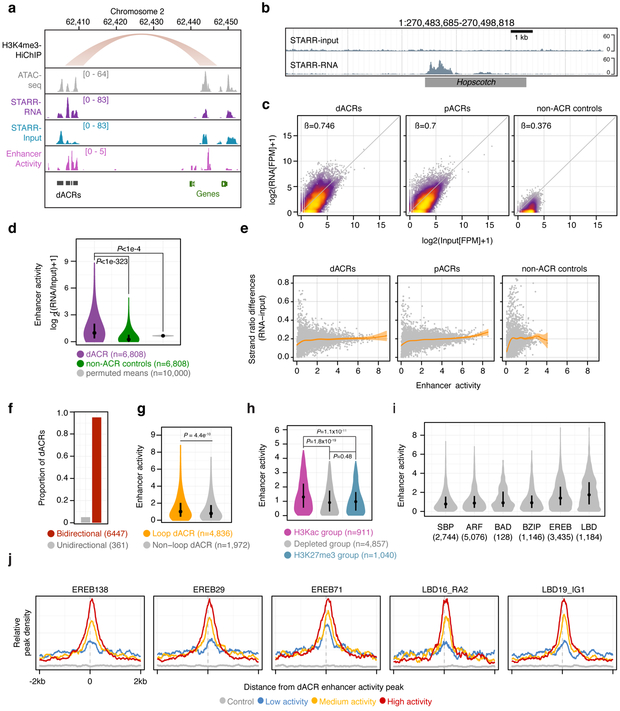
Genetic mapping studies on crops suggest that agronomic traits can be controlled by gene-distal intergenic loci. Despite the biological importance and the potential agronomic utility of these loci, they remain virtually uncharacterized in all crop species to date. Here, we provide genetic, epigenomic and functional molecular evidence to support the widespread existence of gene-distal (hereafter, distal) loci that act as long-range transcriptional cis-regulatory elements (CREs) in the maize genome. Such loci are enriched for euchromatic features that suggest their regulatory functions. Chromatin loops link together putative CREs with genes and recapitulate genetic interactions. Putative CREs also display elevated transcriptional enhancer activities, as measured by self-transcribing active regulatory region sequencing. These results provide functional support for the widespread existence of CREs that act over large genomic distances to control gene expression.
Figures


Comment in
-
From a distance - gene regulation in plants.Nat Rev Genet. 2020 Feb;21(2):68-69. doi: 10.1038/s41576-019-0201-8. Nat Rev Genet. 2020. PMID: 31784717 No abstract available.
References
References Cited in Main Text
-
- Shlyueva D, Stampfel G & Stark A Transcriptional enhancers: from properties to genome-wide predictions. Nat Rev Genet 15, 272–86 (2014). - PubMed
-
- Weber B, Zicola J, Oka R & Stam M Plant Enhancers: A Call for Discovery. Trends Plant Sci 21, 974–987 (2016). - PubMed
-
- Marand AP, Zhang T, Zhu B & Jiang J Towards genome-wide prediction and characterization of enhancers in plants. Biochim Biophys Acta Gene Regul Mech 1860, 131–139 (2017). - PubMed
References Cited in Methods Section Only
-
- Benfey PN & Chua NH The Cauliflower Mosaic Virus 35S Promoter: Combinatorial Regulation of Transcription in Plants. Science 250, 959–66 (1990). - PubMed
-
- Yoo SD, Cho YH & Sheen J Arabidopsis mesophyll protoplasts: a versatile cell system for transient gene expression analysis. Nat Protoc 2, 1565–72 (2007). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
