

Nanopore native RNA sequencing of a human poly(A) transcriptome
- PMID: 31740818
- PMCID: PMC7768885
- DOI: 10.1038/s41592-019-0617-2
Nanopore native RNA sequencing of a human poly(A) transcriptome
Erratum in
-
Author Correction: Nanopore native RNA sequencing of a human poly(A) transcriptome.Nat Methods. 2020 Jan;17(1):114. doi: 10.1038/s41592-019-0697-z. Nat Methods. 2020. PMID: 31819268 Free PMC article.
Abstract
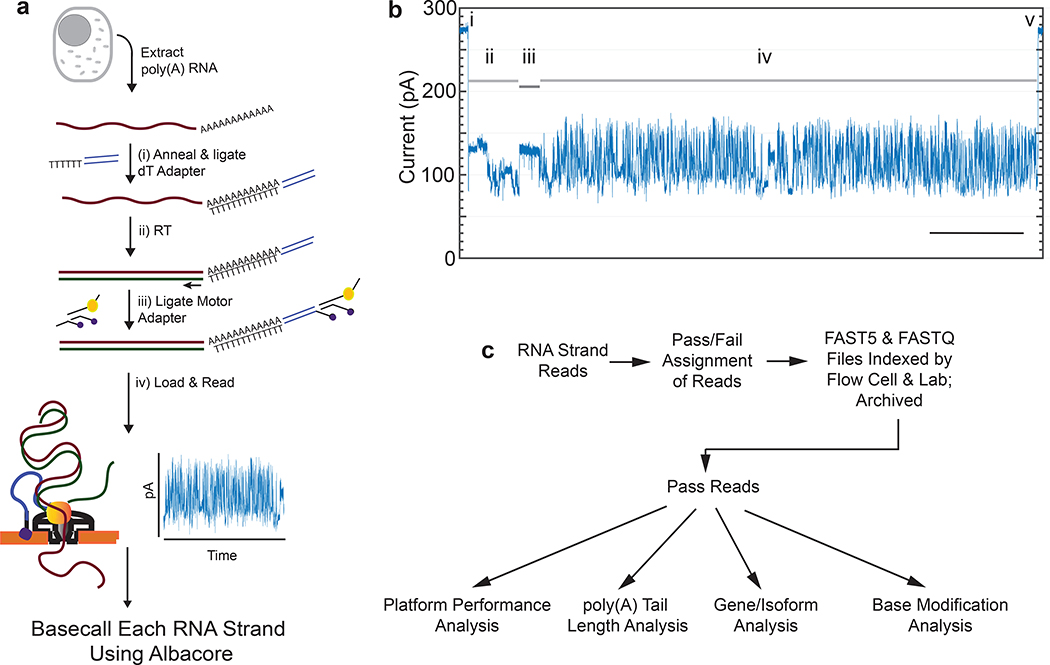
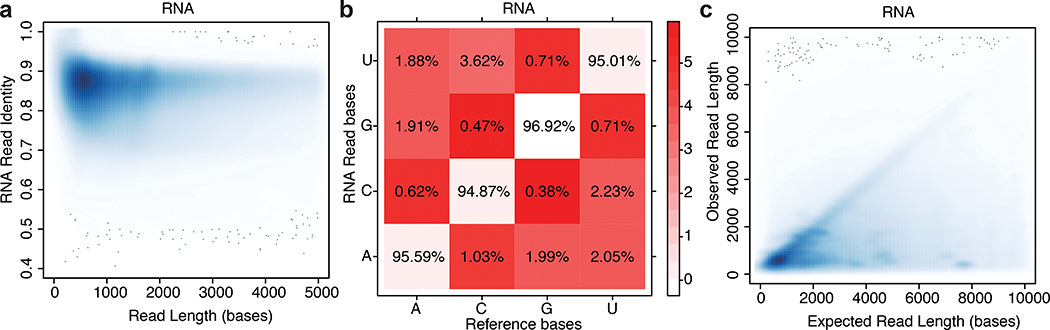
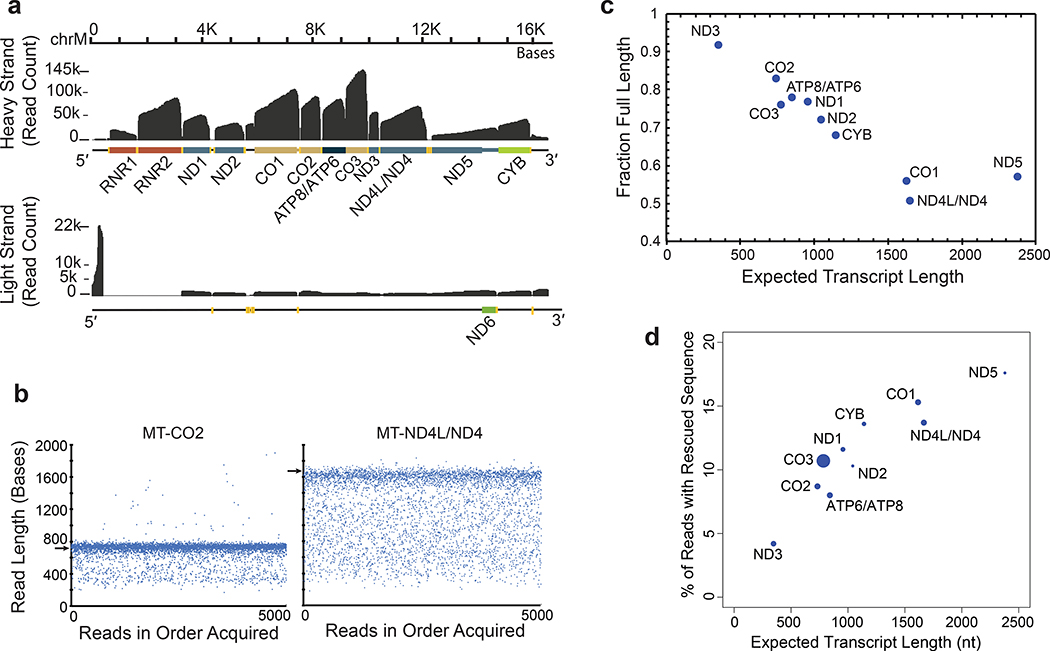
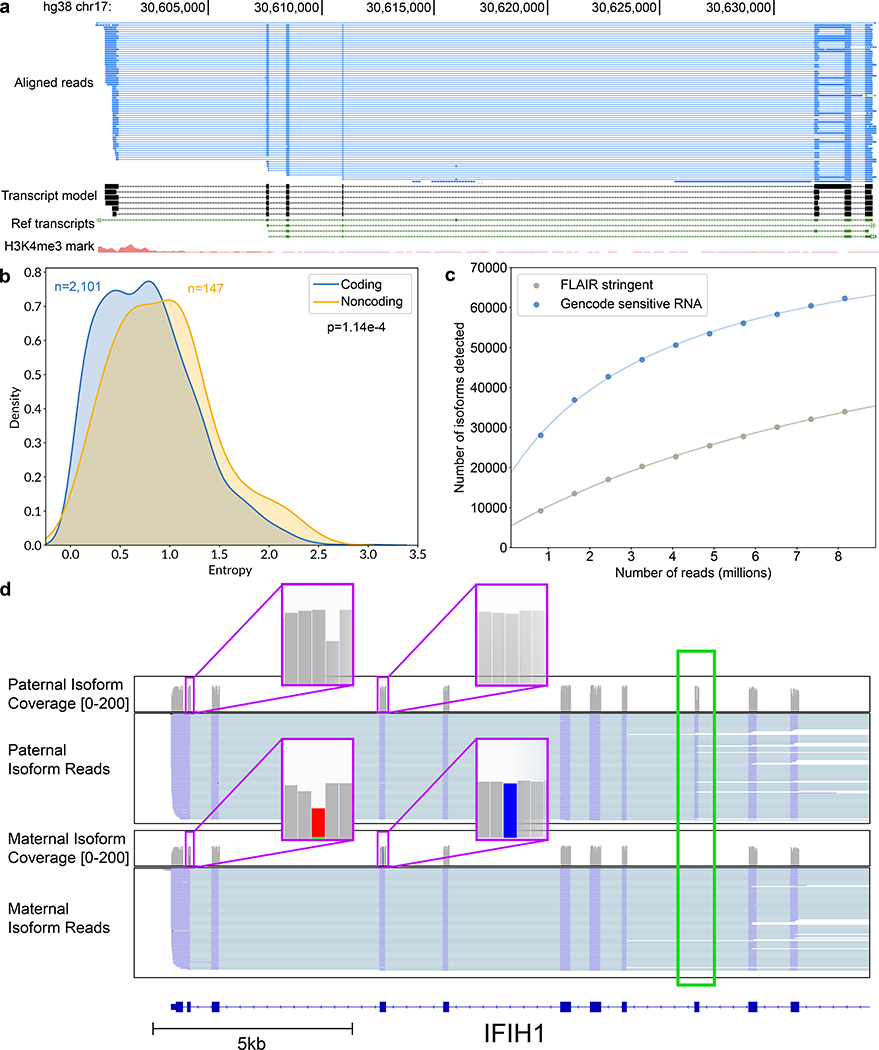
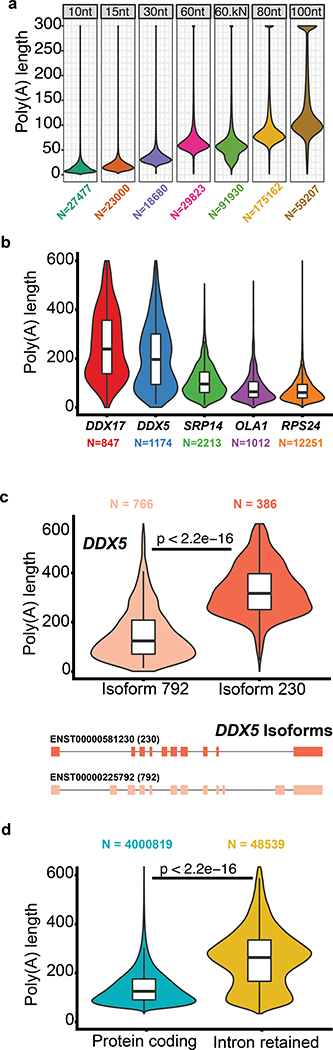
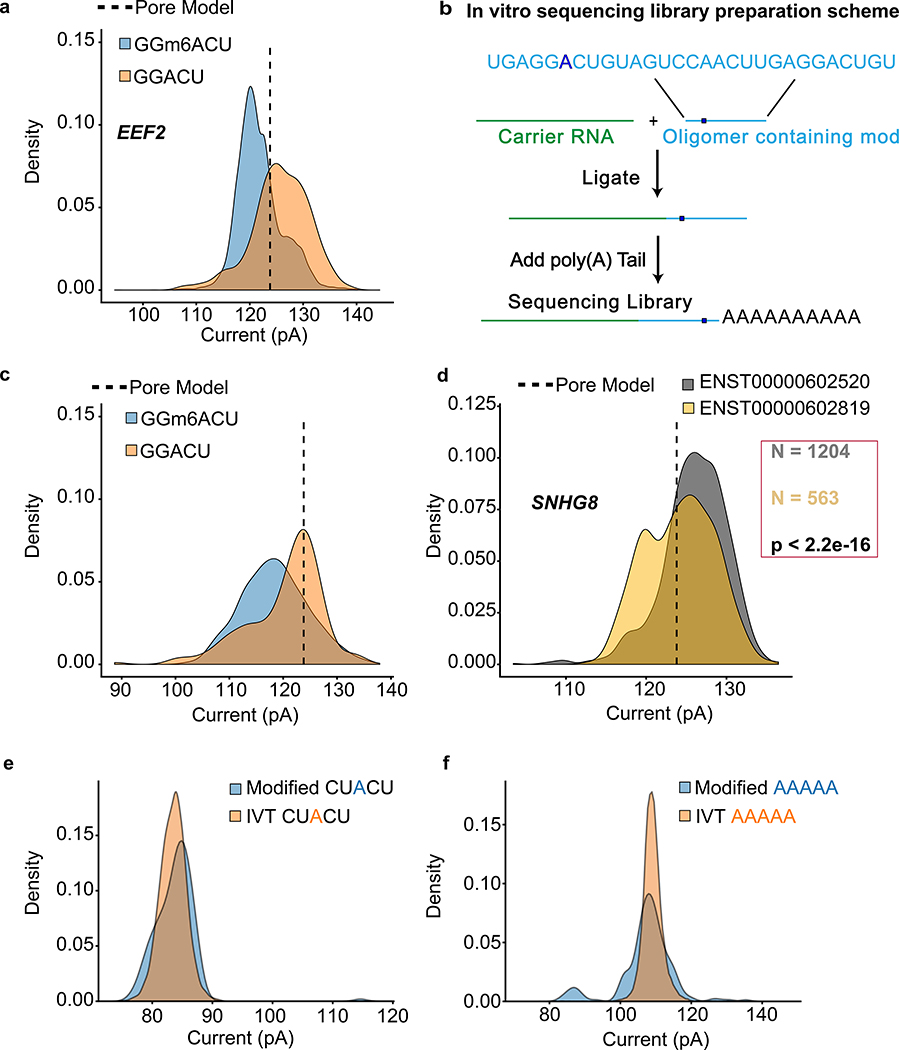
High-throughput complementary DNA sequencing technologies have advanced our understanding of transcriptome complexity and regulation. However, these methods lose information contained in biological RNA because the copied reads are often short and modifications are not retained. We address these limitations using a native poly(A) RNA sequencing strategy developed by Oxford Nanopore Technologies. Our study generated 9.9 million aligned sequence reads for the human cell line GM12878, using thirty MinION flow cells at six institutions. These native RNA reads had a median length of 771 bases, and a maximum aligned length of over 21,000 bases. Mitochondrial poly(A) reads provided an internal measure of read-length quality. We combined these long nanopore reads with higher accuracy short-reads and annotated GM12878 promoter regions to identify 33,984 plausible RNA isoforms. We describe strategies for assessing 3' poly(A) tail length, base modifications and transcript haplotypes.
Conflict of interest statement
COMPETING FINANCIAL INTERESTS
Figures
Comment in
-
Expanding applications for nanopore sequencing.Nat Rev Genet. 2020 Feb;21(2):67. doi: 10.1038/s41576-019-0204-5. Nat Rev Genet. 2020. PMID: 31811281 No abstract available.
References
-
- Complementary DNA sequencing: expressed sequenced tags and human genome project M.D. Adams et al. Science 252, 1651–1656. Trends Genet. 7, 281 (1991). - PubMed
-
- Temin HM & Mizutani S RNA-dependent DNA polymerase in virions of Rous sarcoma virus. Nature 226, 1211–1213 (1970). - PubMed
-
- Baltimore D Viral RNA-dependent DNA Polymerase: RNA-dependent DNA Polymerase in Virions of RNA Tumour Viruses. Nature 226, 1209 (1970). - PubMed
-
- Saiki RK et al. Primer-directed enzymatic amplification of DNA with a thermostable DNA polymerase. Science 239, 487–491 (1988). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
