

Fast, sensitive and accurate integration of single-cell data with Harmony
- PMID: 31740819
- PMCID: PMC6884693
- DOI: 10.1038/s41592-019-0619-0
Fast, sensitive and accurate integration of single-cell data with Harmony
Abstract
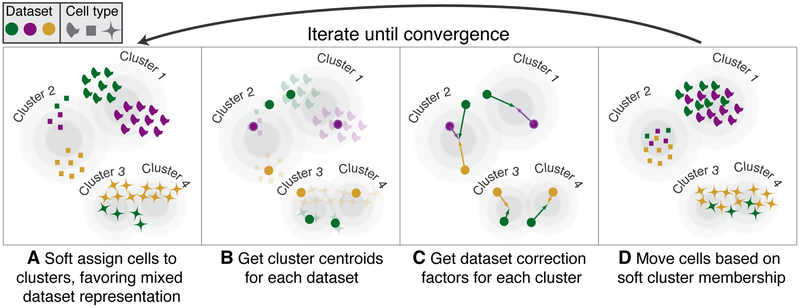
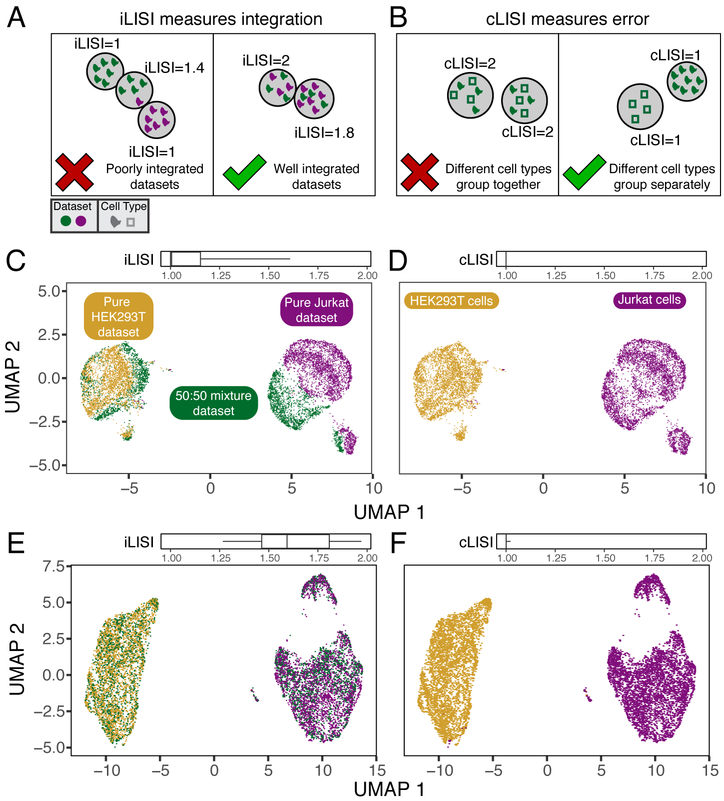
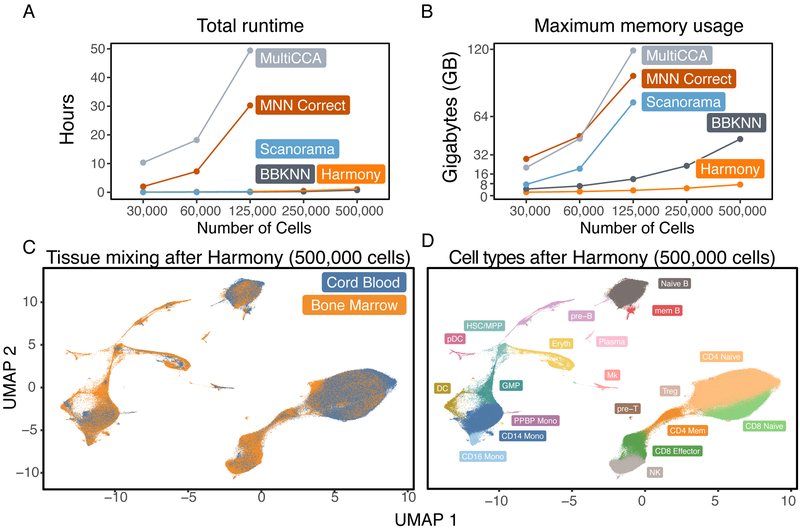
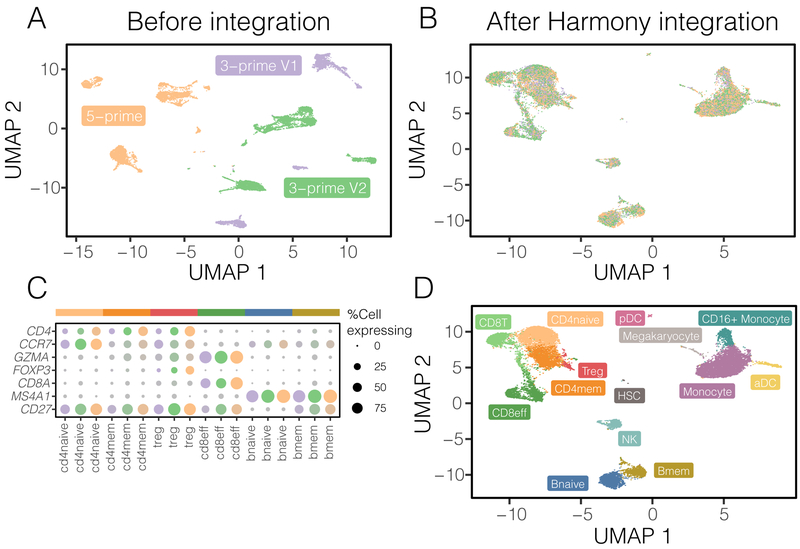
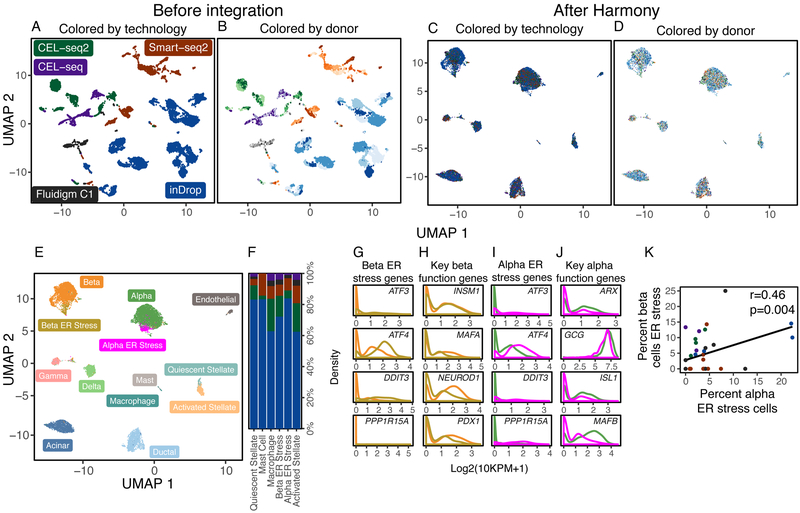
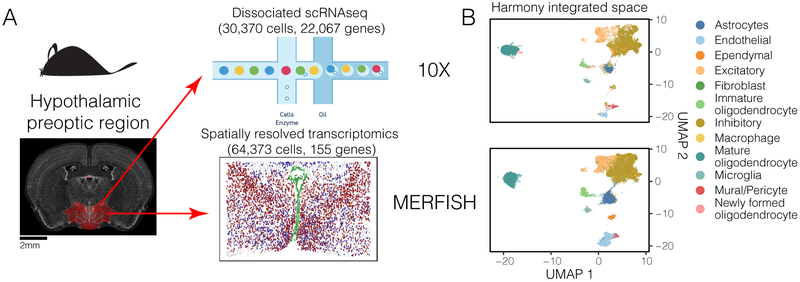
The emerging diversity of single-cell RNA-seq datasets allows for the full transcriptional characterization of cell types across a wide variety of biological and clinical conditions. However, it is challenging to analyze them together, particularly when datasets are assayed with different technologies, because biological and technical differences are interspersed. We present Harmony (https://github.com/immunogenomics/harmony), an algorithm that projects cells into a shared embedding in which cells group by cell type rather than dataset-specific conditions. Harmony simultaneously accounts for multiple experimental and biological factors. In six analyses, we demonstrate the superior performance of Harmony to previously published algorithms while requiring fewer computational resources. Harmony enables the integration of ~106 cells on a personal computer. We apply Harmony to peripheral blood mononuclear cells from datasets with large experimental differences, five studies of pancreatic islet cells, mouse embryogenesis datasets and the integration of scRNA-seq with spatial transcriptomics data.
Figures
References
-
- Svensson V, Vento-Tormo R & Teichmann SA Exponential scaling of single-cell RNA-seq in the past decade. Nature Protocols 13, 599–604 (2018). - PubMed
Methods-only References
-
- Mao Q, Wang L, Goodison S & Sun Y Dimensionality reduction via graph structure learning. In Proceedings of the 21th ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, KDD ‘15, 765–774 (ACM, New York, NY, USA, 2015).
-
- Dhillon IS & Modha DS Concept decompositions for large sparse text data using clustering. Mach. Learn. 42,143–175 (2001).
-
- Jordan MI & Jacobs RA Hierarchical mixtures of experts and the EM algorithm. Neural Comput. 6, 181–214 (1994).
-
- Buttner M, Miao Z, Wolf FA, Teichmann SA & Theis FJ A test metric for assessing single-cell RNA-seq batch correction. Nature Methods 16, 43–49 (2019). - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
