

In situ readout of DNA barcodes and single base edits facilitated by in vitro transcription
- PMID: 31740838
- PMCID: PMC6954335
- DOI: 10.1038/s41587-019-0299-4
In situ readout of DNA barcodes and single base edits facilitated by in vitro transcription
Erratum in
-
Publisher Correction: In situ readout of DNA barcodes and single base edits facilitated by in vitro transcription.Nat Biotechnol. 2020 Feb;38(2):245. doi: 10.1038/s41587-020-0432-4. Nat Biotechnol. 2020. PMID: 31988471
Abstract
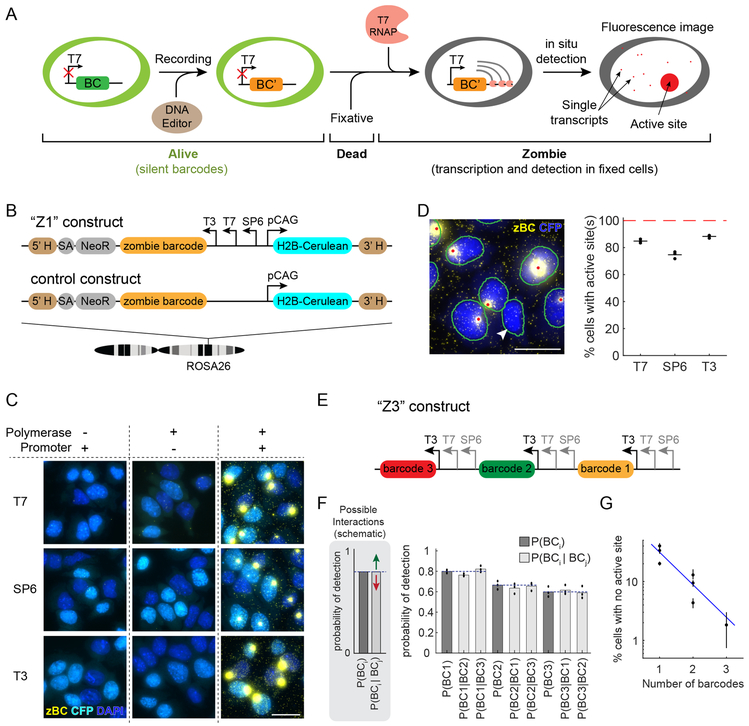
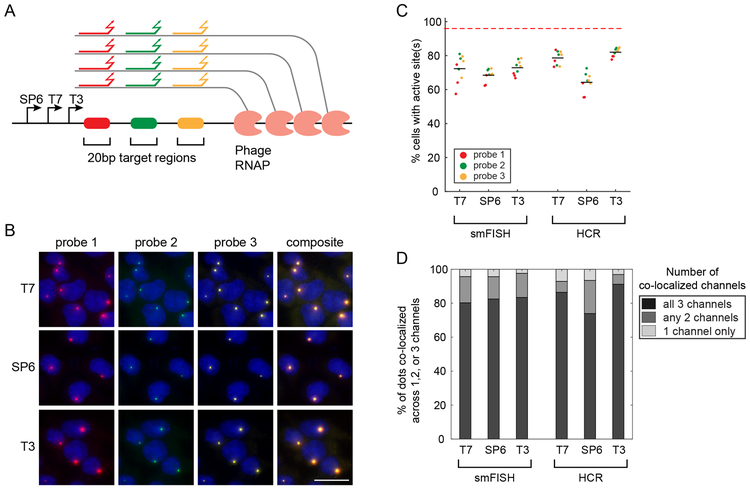
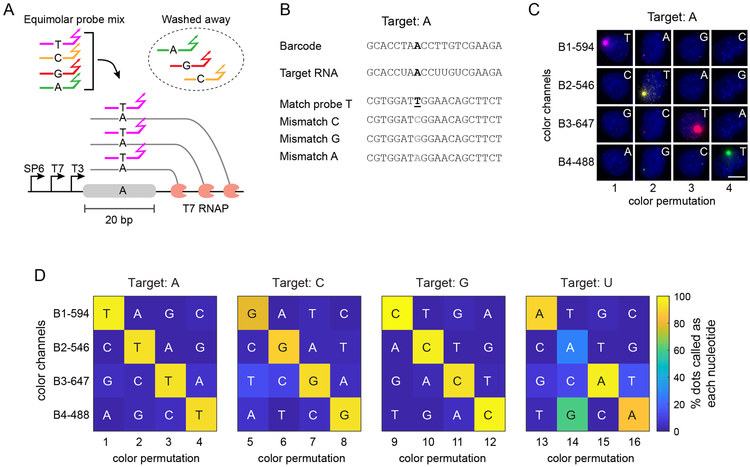
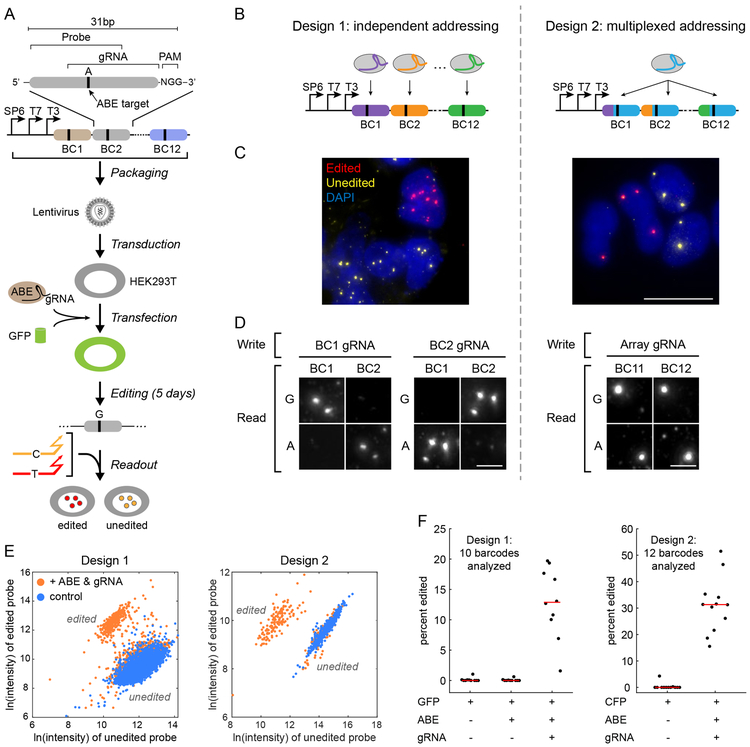
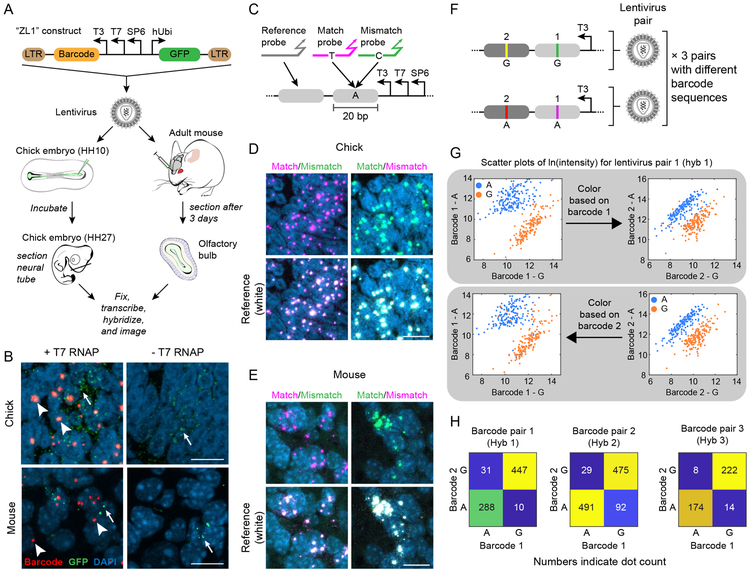
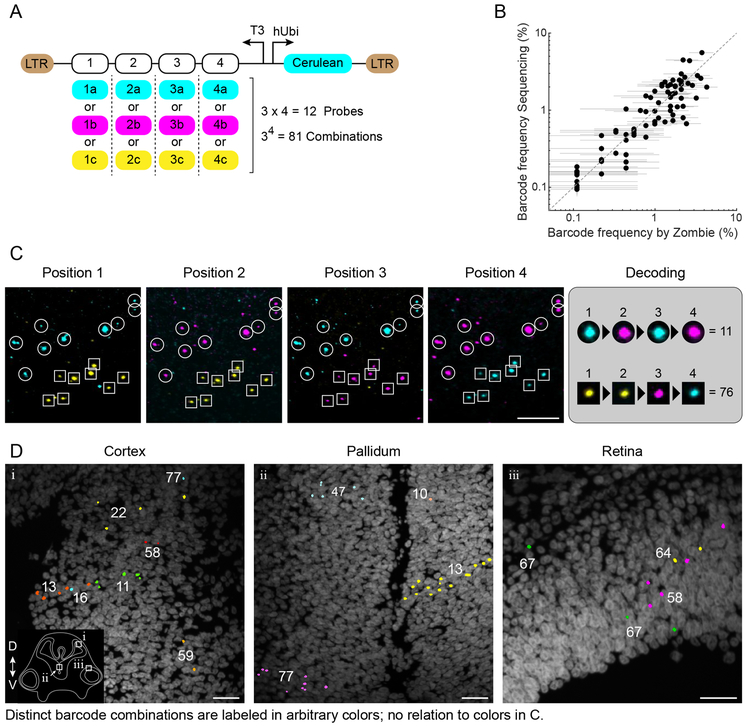
Molecular barcoding technologies that uniquely identify single cells are hampered by limitations in barcode measurement. Readout by sequencing does not preserve the spatial organization of cells in tissues, whereas imaging methods preserve spatial structure but are less sensitive to barcode sequence. Here we introduce a system for image-based readout of short (20-base-pair) DNA barcodes. In this system, called Zombie, phage RNA polymerases transcribe engineered barcodes in fixed cells. The resulting RNA is subsequently detected by fluorescent in situ hybridization. Using competing match and mismatch probes, Zombie can accurately discriminate single-nucleotide differences in the barcodes. This method allows in situ readout of dense combinatorial barcode libraries and single-base mutations produced by CRISPR base editors without requiring barcode expression in live cells. Zombie functions across diverse contexts, including cell culture, chick embryos and adult mouse brain tissue. The ability to sensitively read out compact and diverse DNA barcodes by imaging will facilitate a broad range of barcoding and genomic recording strategies.
Figures
References
-
- Alemany A, Florescu M, Baron CS, Peterson-Maduro J & van Oudenaarden A Whole-organism clone tracing using single-cell sequencing. Nature 556, 108–112 (2018). - PubMed
Methods-only References
-
- Lois C, Hong EJ, Pease S, Brown EJ & Baltimore D Germline transmission and tissue-specific expression of transgenes delivered by lentiviral vectors. Science 295, 868–872 (2002). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
