

A mutation in mannose-phosphate-dolichol utilization defect 1 reveals clinical symptoms of congenital disorders of glycosylation type I and dystroglycanopathy
- PMID: 31741824
- PMCID: PMC6850978
- DOI: 10.1002/jmd2.12060
A mutation in mannose-phosphate-dolichol utilization defect 1 reveals clinical symptoms of congenital disorders of glycosylation type I and dystroglycanopathy
Abstract
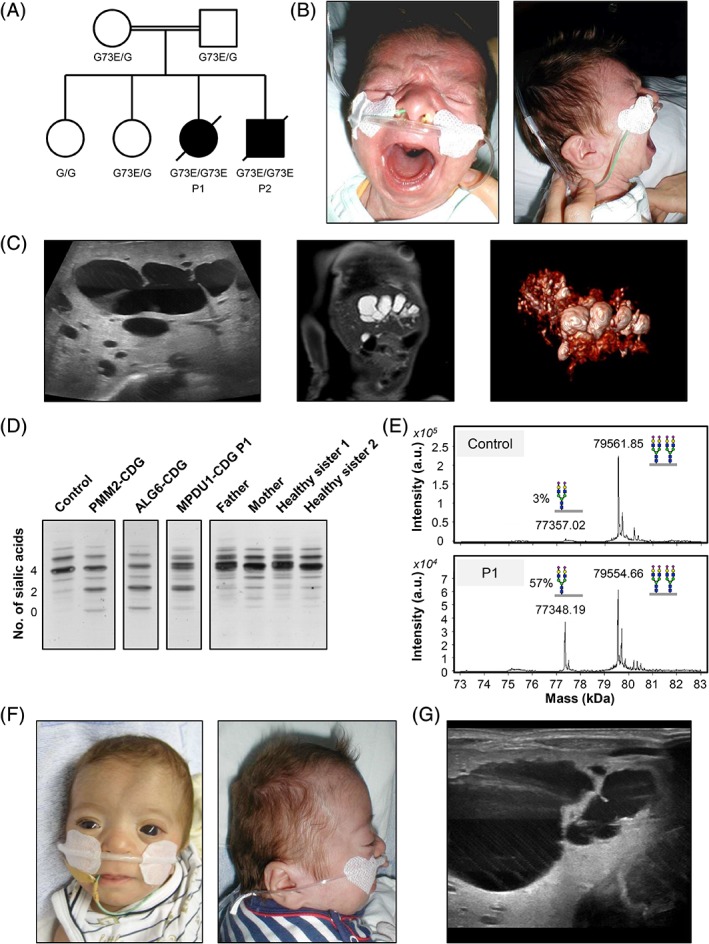
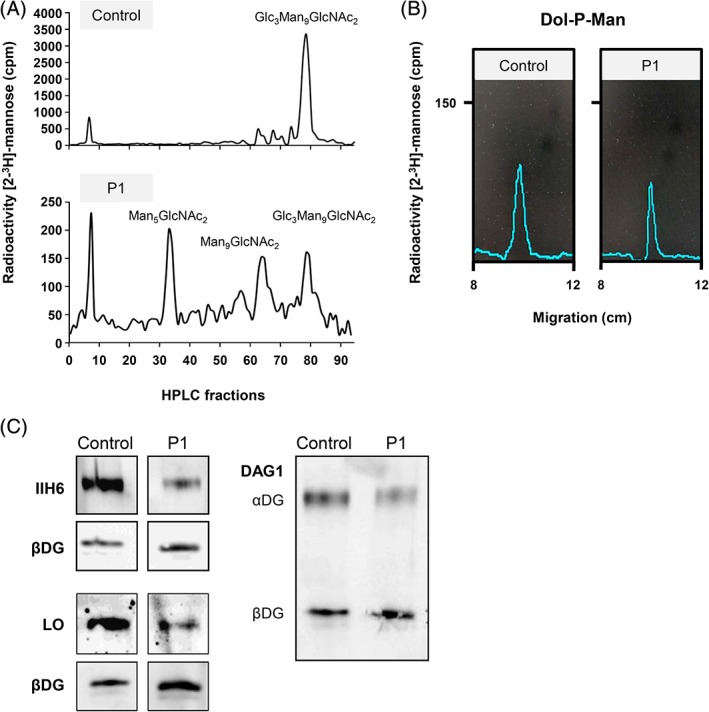
Congenital disorders of glycosylation type I (CDG-I) are inborn errors of metabolism, generally characterized by multisystem clinical manifestations, including developmental delay, hepatopathy, hypotonia, and skin, skeletal, and neurological abnormalities. Among others, dolichol-phosphate-mannose (DPM) is the mannose donor for N-glycosylation as well as O-mannosylation. DOLK-CDG, DPM1-CDG, DPM2-CDG, and DPM3-CDG are defects in the DPM synthesis showing both CDG-I abnormalities and reduced O-mannosylation of alpha-dystroglycan (αDG), which leads to muscular dystrophy-dystroglycanopathy. Mannose-phosphate-dolichol utilization defect 1 (MPDU1) plays a role in the utilization of DPM. Here, we report two MPDU1-CDG patients without skin involvement, but with massive dilatation of the biliary duct system and dystroglycanopathy characteristics including hypotonia, elevated creatine kinase, dilated cardiomyopathy, buphthalmos, and congenital glaucoma. Biochemical analyses revealed elevated disialotransferrin in serum, and analyses in fibroblasts showed shortened lipid linked oligosaccharides and DPM, and reduced O-mannosylation of αDG. Thus, MPDU1-CDG can be added to the list of disorders with overlapping biochemical and clinical abnormalities of CDG-I and dystroglycanopathy.
Synopsis: Mannose-phosphate-dolichol utilization defect 1 patients can have overlapping biochemical and clinical abnormalities of congenital disorders of glycosylation type I and dystroglycanopathy.
Keywords: MPDU1‐CDG; congenital disorders of glycosylation; dolichol‐phosphate‐mannose; dystroglycanopathy.
© 2019 The Authors. Journal of Inherited Metabolic Disease published by John Wiley & Sons Ltd on behalf of SSIEM.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures
References
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
