

ADAPTS: Automated deconvolution augmentation of profiles for tissue specific cells
- PMID: 31743345
- PMCID: PMC6863530
- DOI: 10.1371/journal.pone.0224693
ADAPTS: Automated deconvolution augmentation of profiles for tissue specific cells
Abstract
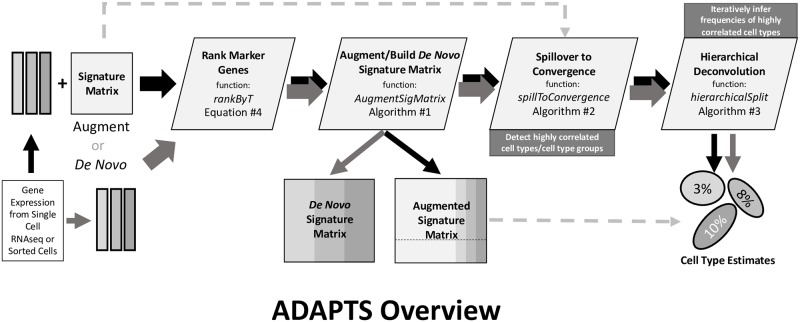
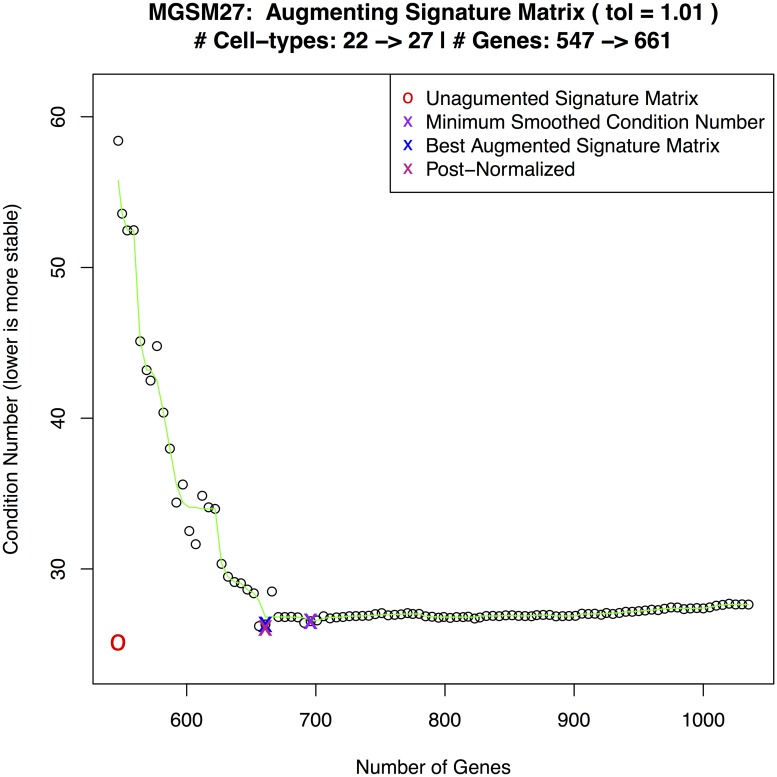
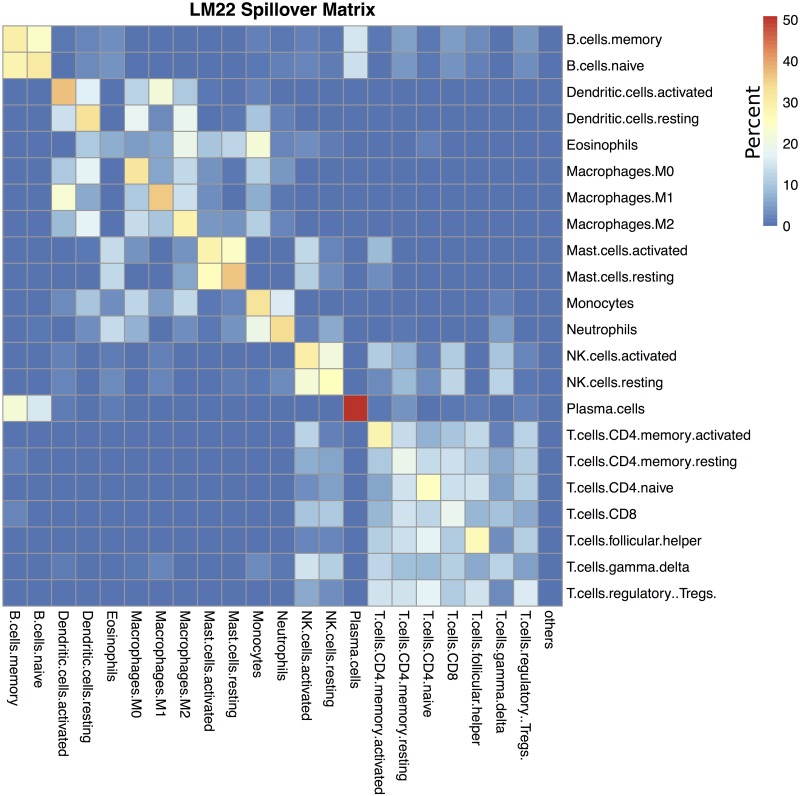
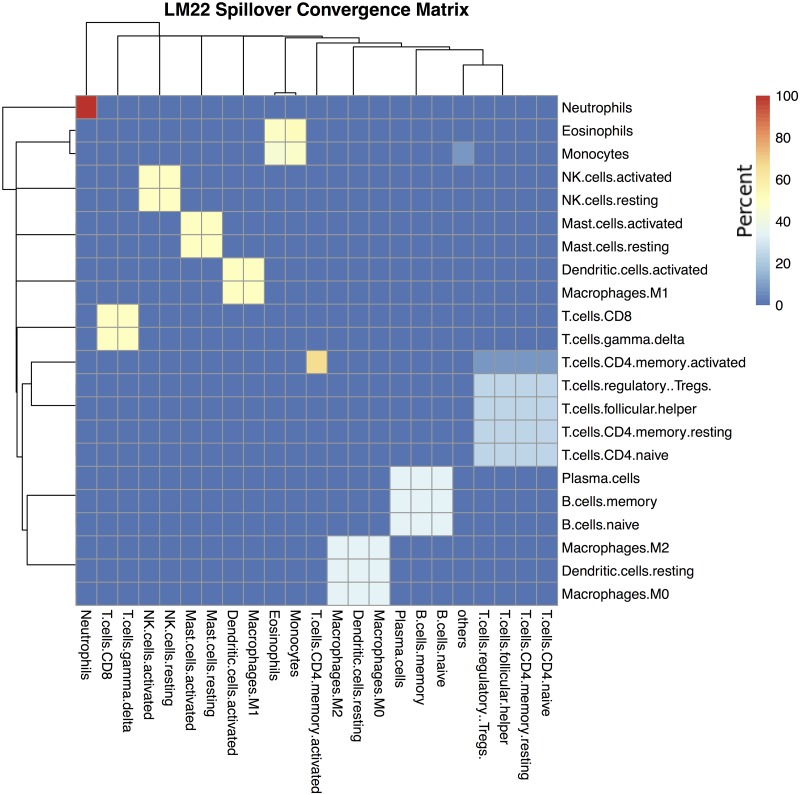
Immune cell infiltration of tumors and the tumor microenvironment can be an important component for determining patient outcomes. For example, immune and stromal cell presence inferred by deconvolving patient gene expression data may help identify high risk patients or suggest a course of treatment. One particularly powerful family of deconvolution techniques uses signature matrices of genes that uniquely identify each cell type as determined from single cell type purified gene expression data. Many methods from this family have been recently published, often including new signature matrices appropriate for a single purpose, such as investigating a specific type of tumor. The package ADAPTS helps users make the most of this expanding knowledge base by introducing a framework for cell type deconvolution. ADAPTS implements modular tools for customizing signature matrices for new tissue types by adding custom cell types or building new matrices de novo, including from single cell RNAseq data. It includes a common interface to several popular deconvolution algorithms that use a signature matrix to estimate the proportion of cell types present in heterogenous samples. ADAPTS also implements a novel method for clustering cell types into groups that are difficult to distinguish by deconvolution and then re-splitting those clusters using hierarchical deconvolution. We demonstrate that the techniques implemented in ADAPTS improve the ability to reconstruct the cell types present in a single cell RNAseq data set in a blind predictive analysis. ADAPTS is currently available for use in R on CRAN and GitHub.
Conflict of interest statement
Our commercial affiliation with the Institute for Systems Biology and Celgene Corporation has no other relevant declarations relating to employment, consultancy, patents, products in development, or marketed products. No author has any competing interest that interferes with, or could reasonably be perceived as interfering with, the full and objective presentation, peer review, editorial decision-making, or publication of research or non-research articles submitted to this journal. This (commercial affiliation) does not alter our adherence to PLOS ONE policies on sharing data and materials.
Figures
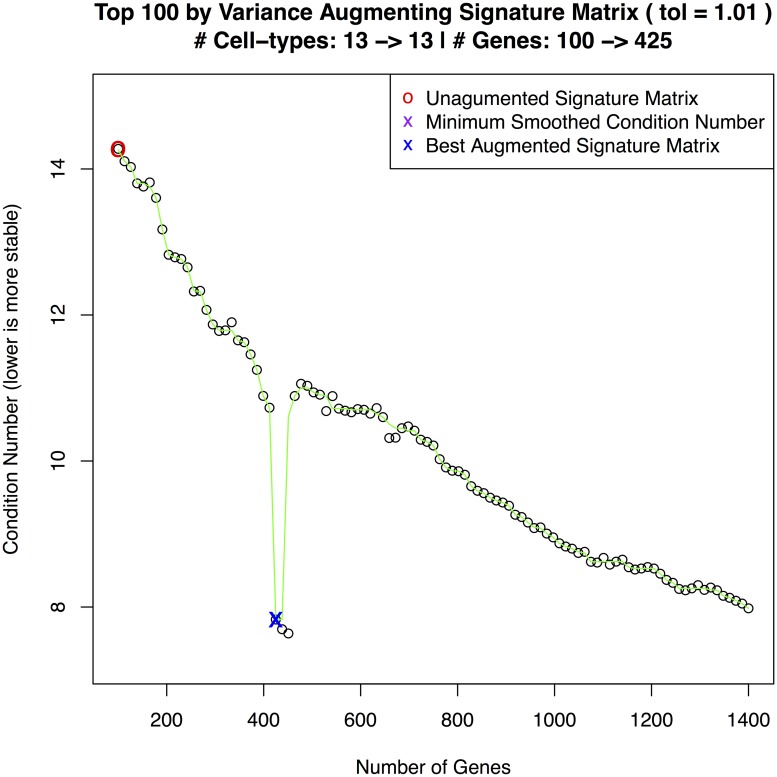
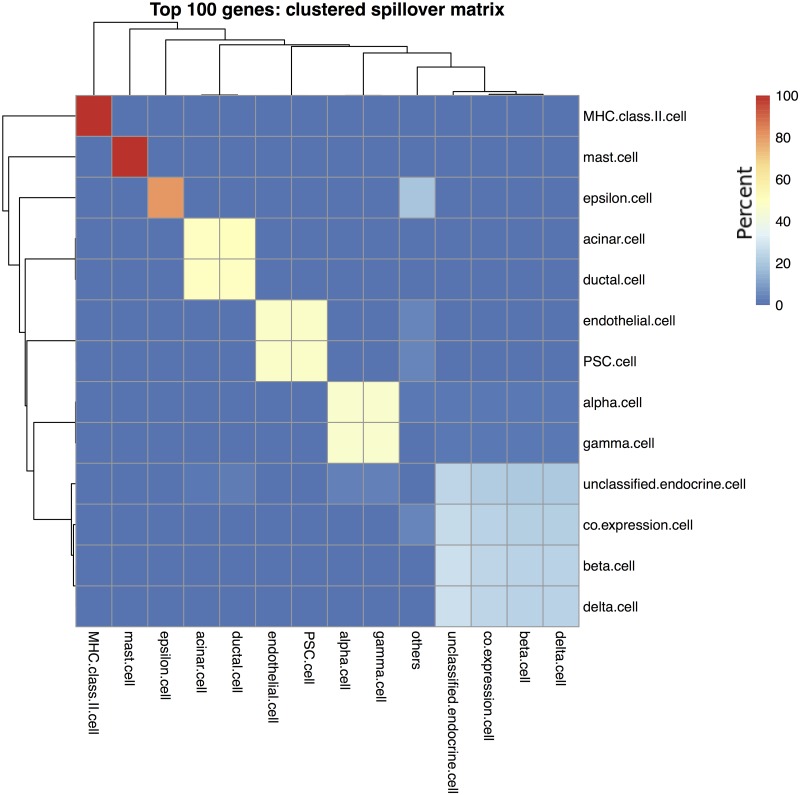
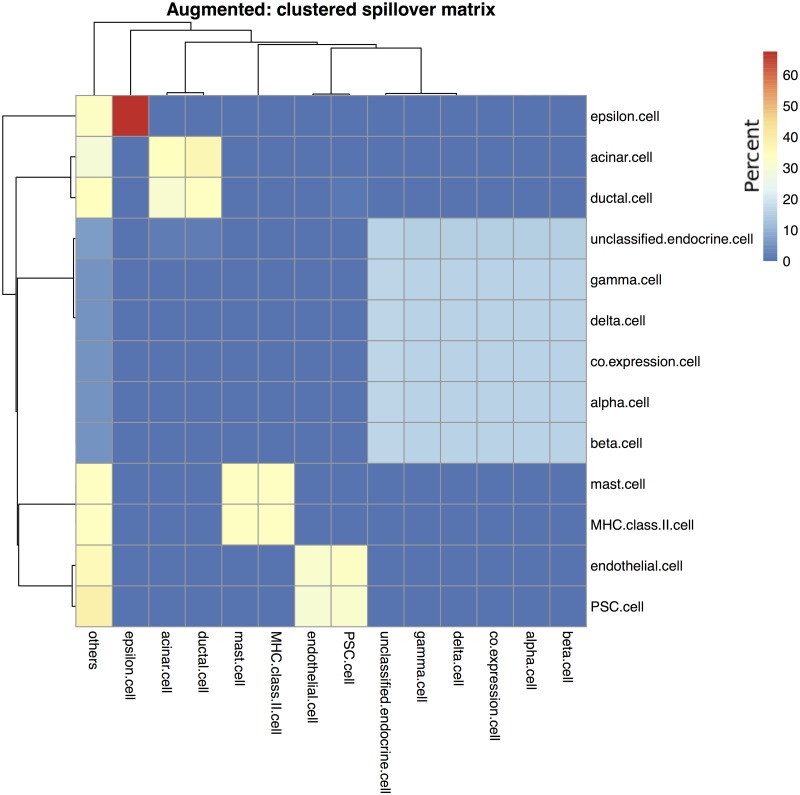
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
