

Endothelial Cell-Derived Interleukin-18 Released During Ischemia Reperfusion Injury Selectively Expands T Peripheral Helper Cells to Promote Alloantibody Production
- PMID: 31744330
- PMCID: PMC7035199
- DOI: 10.1161/CIRCULATIONAHA.119.042501
Endothelial Cell-Derived Interleukin-18 Released During Ischemia Reperfusion Injury Selectively Expands T Peripheral Helper Cells to Promote Alloantibody Production
Abstract
Background: Ischemia reperfusion injury (IRI) predisposes to the formation of donor-specific antibodies, a factor contributing to chronic rejection and late allograft loss.
Methods: We describe a mechanism underlying the correlative association between IRI and donor-specific antibodies by using humanized models and patient specimens.
Results: IRI induces immunoglobulin M-dependent complement activation on endothelial cells that assembles an NLRP3 (NOD-like receptor pyrin domain-containing protein 3) inflammasome via a Rab5-ZFYVE21-NIK axis and upregulates ICOS-L (inducible costimulator ligand) and PD-L2 (programmed death ligand 2). Endothelial cell-derived interleukin-18 (IL-18) selectively expands a T-cell population (CD4+CD45RO+PD-1hiICOS+CCR2+CXCR5-) displaying features of recently described T peripheral helper cells. This population highly expressed IL-18R1 and promoted donor-specific antibodies in response to IL-18 in vivo. In patients with delayed graft function, a clinical manifestation of IRI, these cells were Ki-67+IL-18R1+ and could be expanded ex vivo in response to IL-18.
Conclusions: IRI promotes elaboration of IL-18 from endothelial cells to selectively expand alloreactive IL-18R1+ T peripheral helper cells in allograft tissues to promote donor-specific antibody formation.
Keywords: T-lymphocytes, helper-inducer; antibody specificity; complement system proteins; inflammasomes; interleukin-18; reperfusion injury.
Conflict of interest statement
CONFLICT OF INTEREST DISCLOSURES
All of the authors listed in this manuscript declare no financial conflicts of interest and have no financial disclosures.
Figures
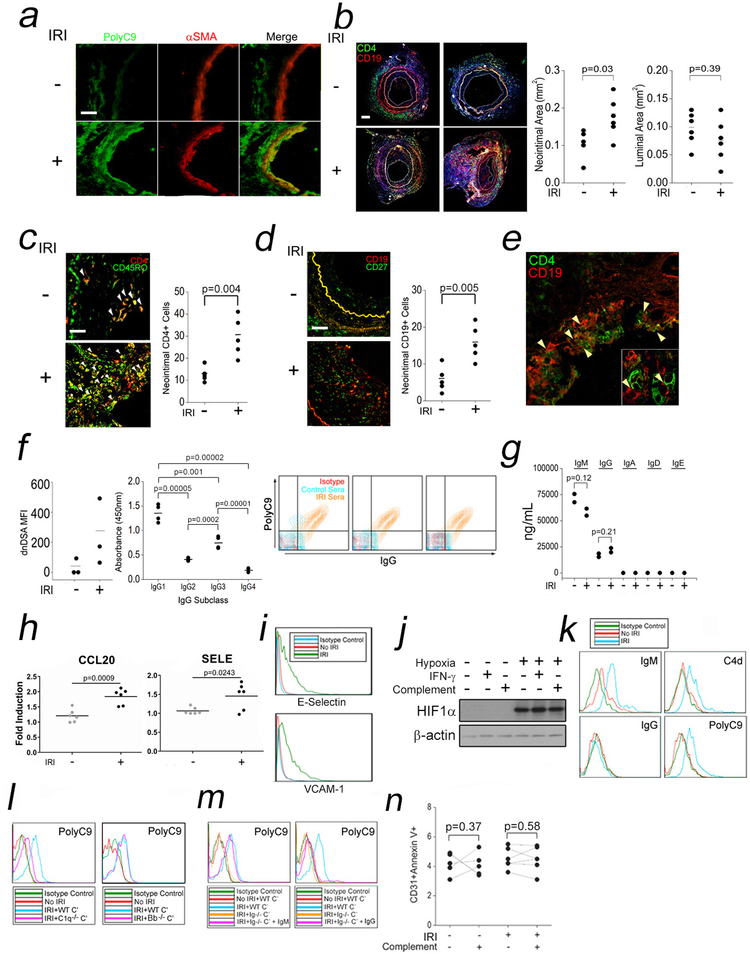



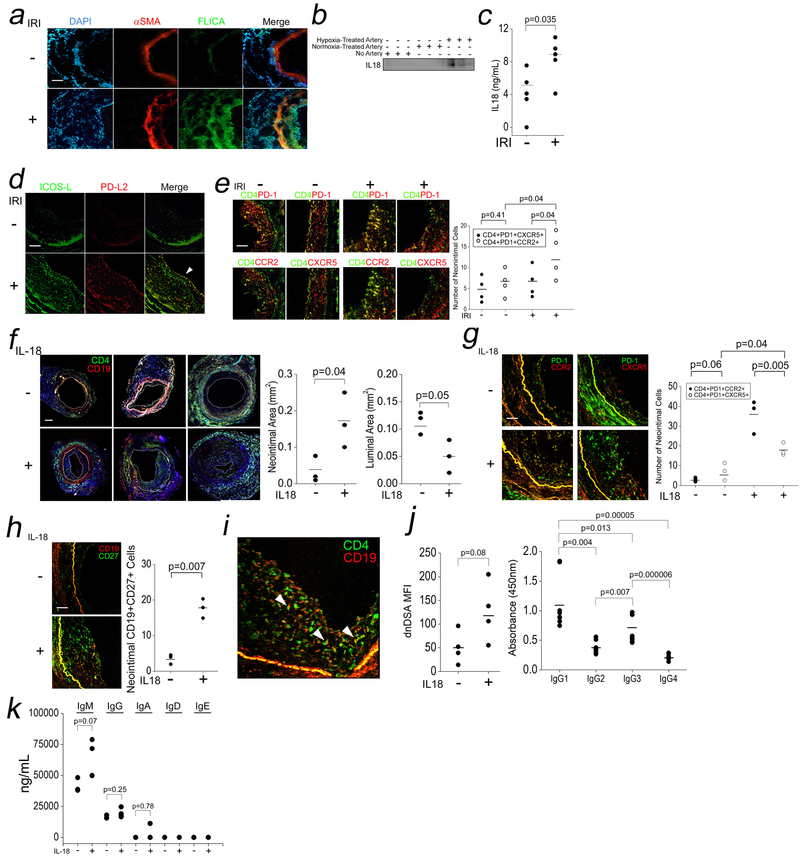
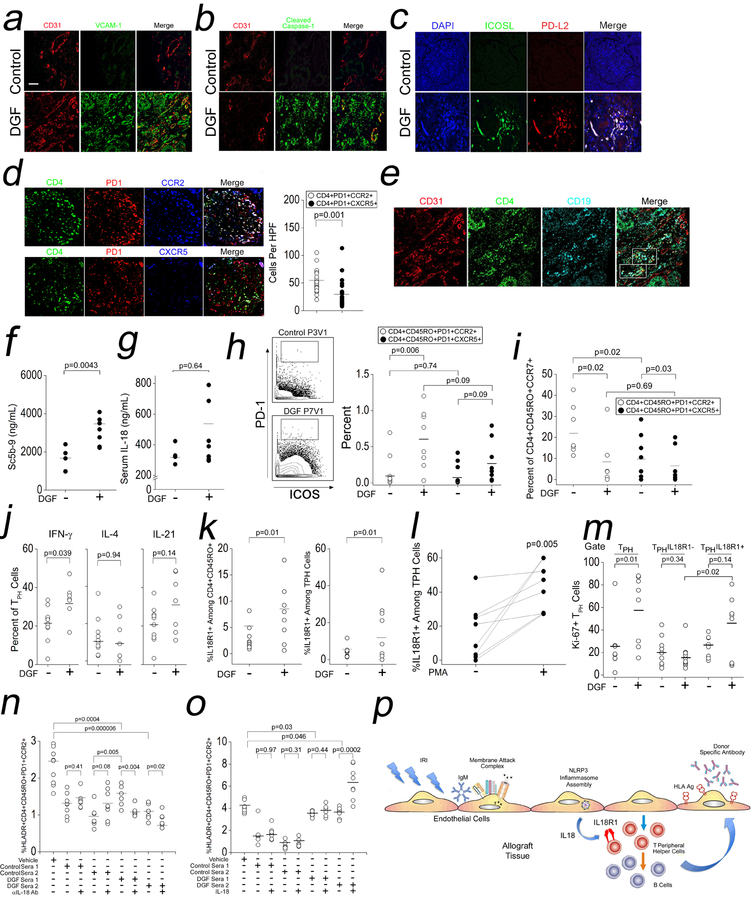
Comment in
-
Mechanisms Underlying Antibody-Mediated Rejection.Circulation. 2020 Feb 11;141(6):479-481. doi: 10.1161/CIRCULATIONAHA.119.044541. Epub 2020 Feb 10. Circulation. 2020. PMID: 32078432 Free PMC article. No abstract available.
References
-
- Wu WK, Famure O, Li Y, Kim SJ. Delayed graft function and the risk of acute rejection in the modern era of kidney transplantation. Kidney Int. 2015;88:851–858. - PubMed
-
- Levine DJ, Glanville AR, Aboyoun C, Belperio J, Benden C, Berry GJ, Hachem R, Hayes D Jr, Neil D, Reinsmoen NL, et al. Antibody-mediated rejection of the lung: a consensus report of the International Society for Heart and Lung Transplantation. J Heart Lung Transplant. 2016; 35:397–406. - PubMed
-
- Demetris AJ, Bellamy C, Hubscher SG, O’Leary J, Randhawa PS, Feng S, Neil D, Colvin RB, McCaughan G, Fung JJ, et al. Comprehensive update of the Banff Working Group on Liver Allograft Pathology: introduction of antibody-mediated rejection. Am J Transplant. 2016;16:2816–2835. - PubMed
-
- Haas M, Loupy A, Lefaucheur C, Roufosse C, Glotz D, Seron D, Nankivell BJ, Halloran PF, Colvin RB, Akalin E, et al. The Banff 2017 Kidney Meeting Report: revised diagnostic criteria for chronic active T cell-mediated rejection, antibody-mediated rejection, and prospects for integrative endpoints for next-generation clinical trials. Am J Transplant. 2018;18:293–307. - PMC - PubMed
-
- Wiebe C, Gibson IW, Blydt-Hansen TD, Karpinski M, Ho J, Storsley LJ, Goldberg A, Birk PE, Rush DN, Nickerson PW. Evolution and clinical pathologic correlations of de novo donor-specific HLA antibody post kidney transplant. Am J Transplant. 2012;12:1157–1167. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
