

Transposable element expression in tumors is associated with immune infiltration and increased antigenicity
- PMID: 31745090
- PMCID: PMC6864081
- DOI: 10.1038/s41467-019-13035-2
Transposable element expression in tumors is associated with immune infiltration and increased antigenicity
Abstract
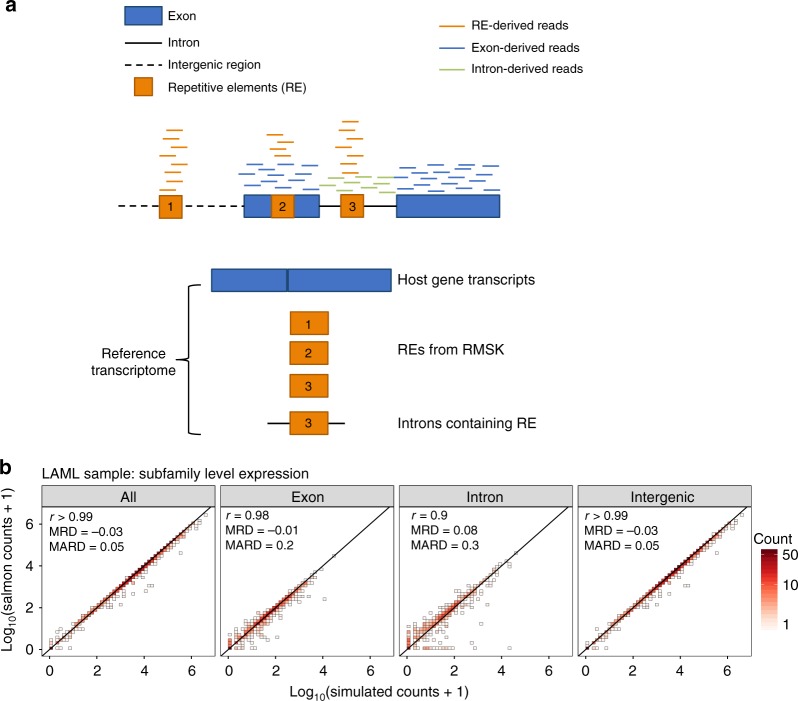
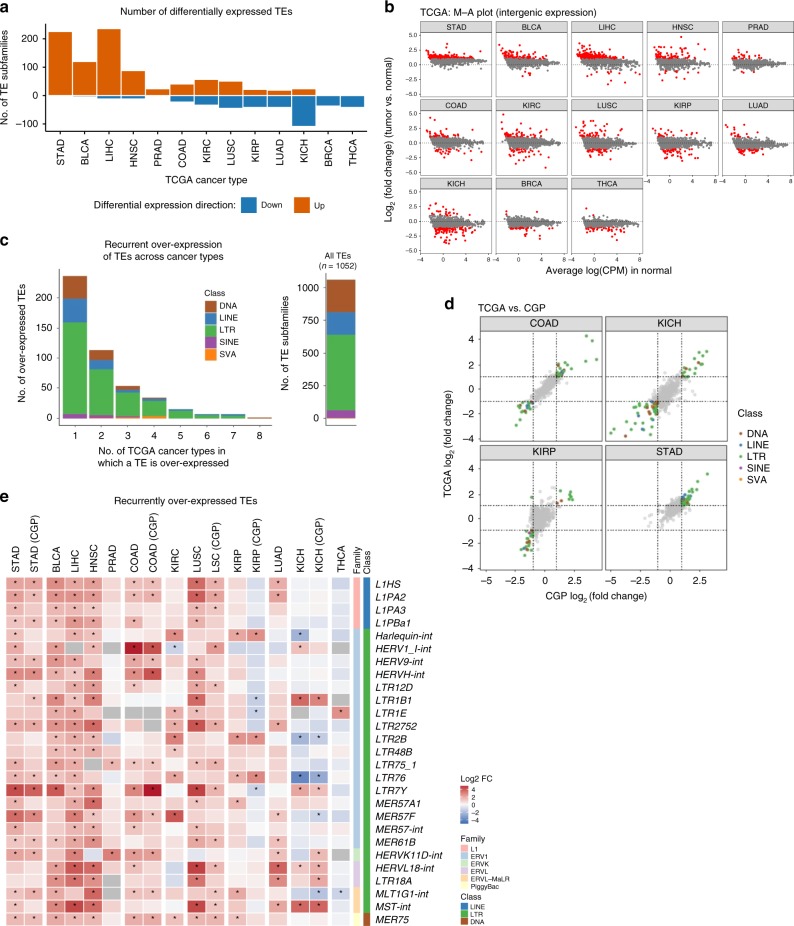
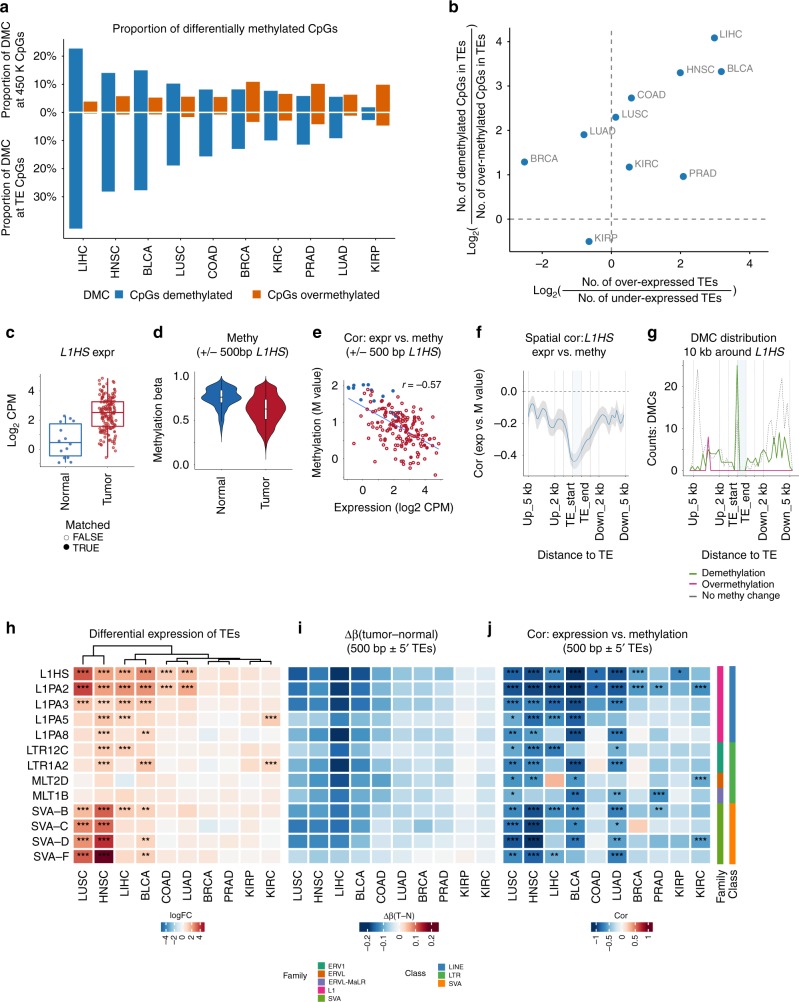
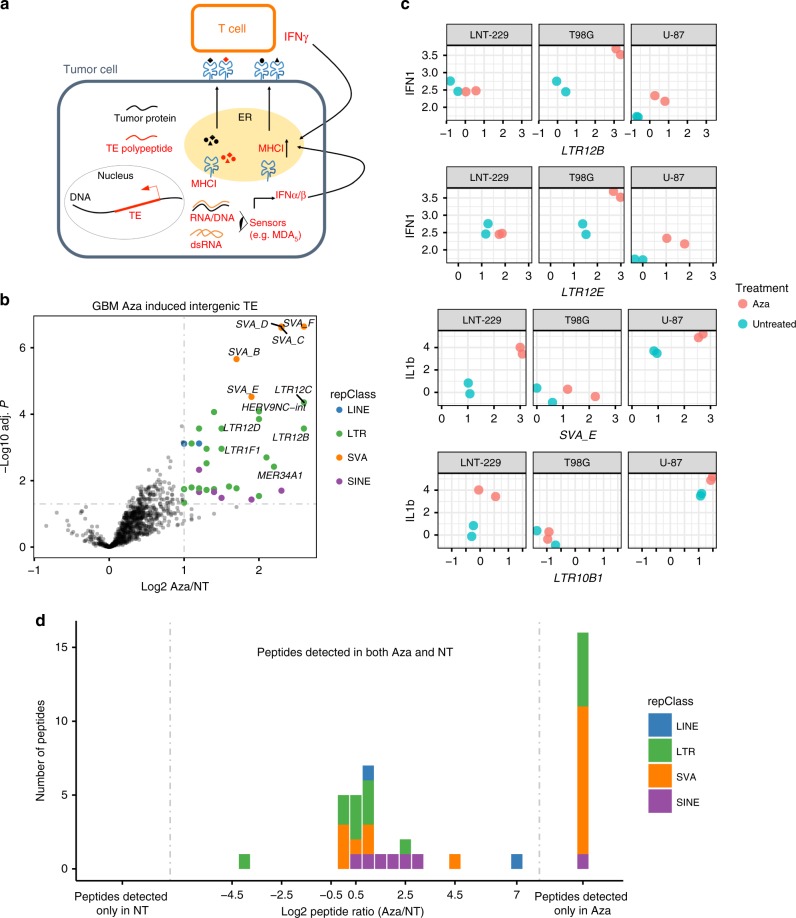
Profound global loss of DNA methylation is a hallmark of many cancers. One potential consequence of this is the reactivation of transposable elements (TEs) which could stimulate the immune system via cell-intrinsic antiviral responses. Here, we develop REdiscoverTE, a computational method for quantifying genome-wide TE expression in RNA sequencing data. Using The Cancer Genome Atlas database, we observe increased expression of over 400 TE subfamilies, of which 262 appear to result from a proximal loss of DNA methylation. The most recurrent TEs are among the evolutionarily youngest in the genome, predominantly expressed from intergenic loci, and associated with antiviral or DNA damage responses. Treatment of glioblastoma cells with a demethylation agent results in both increased TE expression and de novo presentation of TE-derived peptides on MHC class I molecules. Therapeutic reactivation of tumor-specific TEs may synergize with immunotherapy by inducing inflammation and the display of potentially immunogenic neoantigens.
Conflict of interest statement
C.M.R., M.D., A.-J.T., C.B., I.M., R.B., S.J. are employees of Genentech. A.G.W., S.L., M.L.A., P.M.H., H.C.-H. were employees of Genentech. H.C.-H. is the founder of Argonaut Genomics, Inc. The remaining authors declare no competing interests.
Figures

References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
