

Analyses of inter-individual variations of sperm DNA methylation and their potential implications in cattle
- PMID: 31752687
- PMCID: PMC6873545
- DOI: 10.1186/s12864-019-6228-6
Analyses of inter-individual variations of sperm DNA methylation and their potential implications in cattle
Abstract
Background: DNA methylation has been shown to be involved in many biological processes, including X chromosome inactivation in females, paternal genomic imprinting, and others.
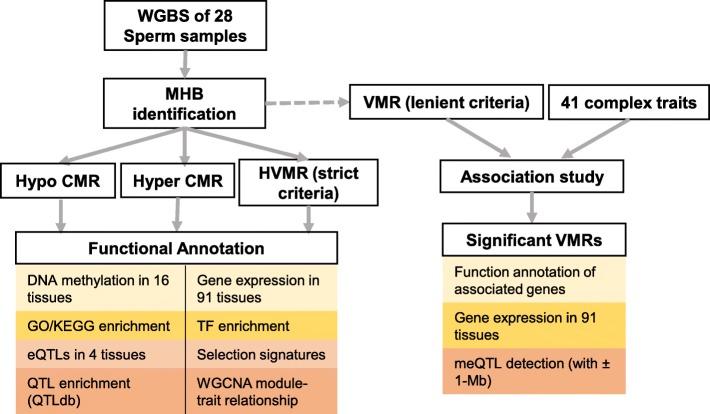
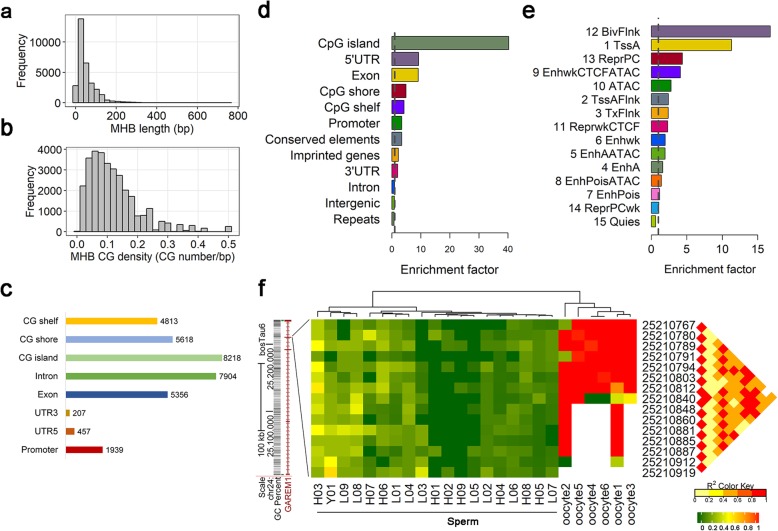
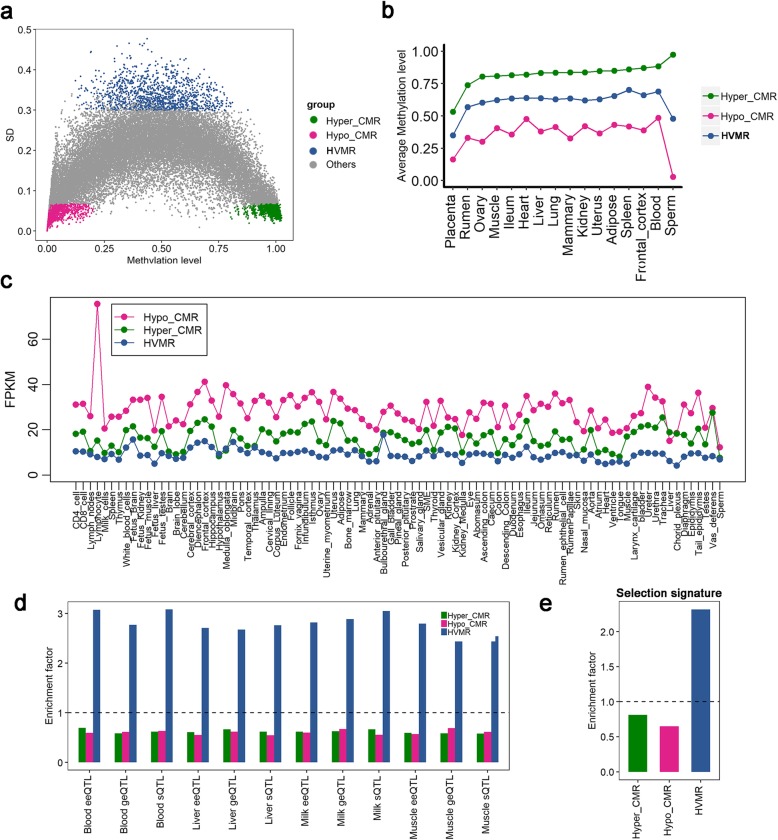
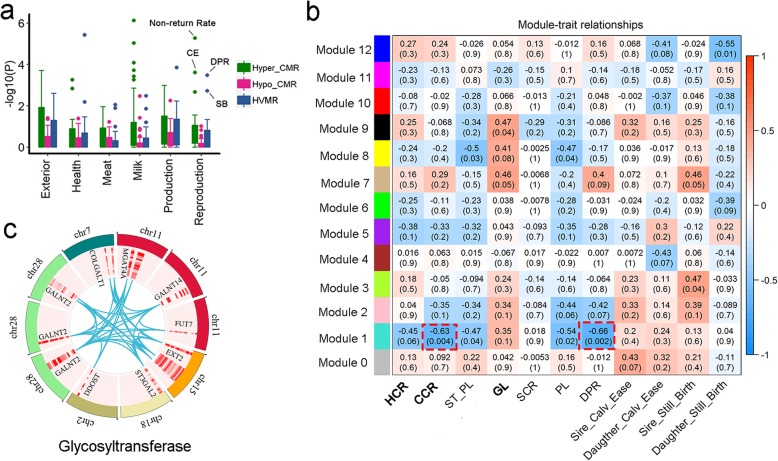
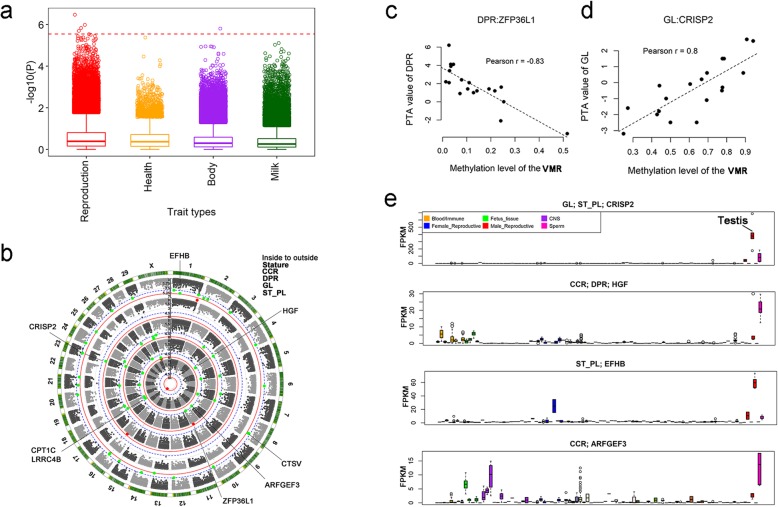
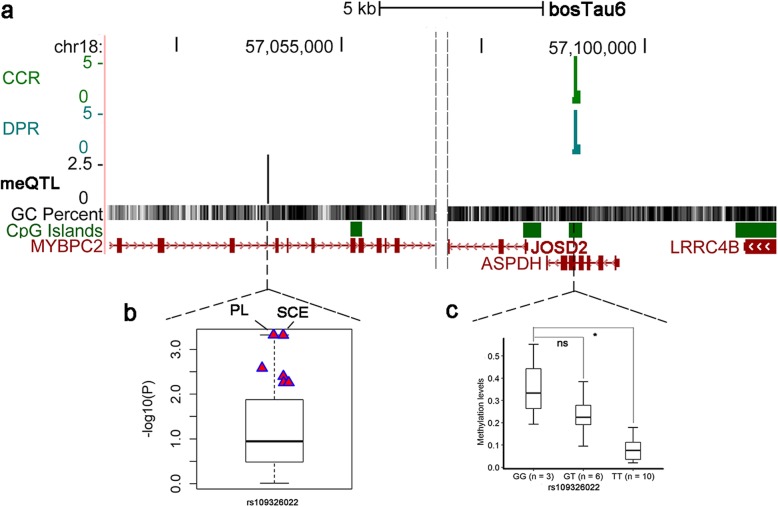
Results: Based on the correlation patterns of methylation levels of neighboring CpG sites among 28 sperm whole genome bisulfite sequencing (WGBS) data (486 × coverage), we obtained 31,272 methylation haplotype blocks (MHBs). Among them, we defined conserved methylated regions (CMRs), variably methylated regions (VMRs) and highly variably methylated regions (HVMRs) among individuals, and showed that HVMRs might play roles in transcriptional regulation and function in complex traits variation and adaptive evolution by integrating evidence from traditional and molecular quantitative trait loci (QTL), and selection signatures. Using a weighted correlation network analysis (WGCNA), we also detected a co-regulated module of HVMRs that was significantly associated with reproduction traits, and enriched for glycosyltransferase genes, which play critical roles in spermatogenesis and fertilization. Additionally, we identified 46 VMRs significantly associated with reproduction traits, nine of which were regulated by cis-SNPs, implying the possible intrinsic relationships among genomic variations, DNA methylation, and phenotypes. These significant VMRs were co-localized (± 10 kb) with genes related to sperm motility and reproduction, including ZFP36L1, CRISP2 and HGF. We provided further evidence that rs109326022 within a predominant QTL on BTA18 might influence the reproduction traits through regulating the methylation level of nearby genes JOSD2 and ASPDH in sperm.
Conclusion: In summary, our results demonstrated associations of sperm DNA methylation with reproduction traits, highlighting the potential of epigenomic information in genomic improvement programs for cattle.
Keywords: Cattle; Methylation haplotype blocks; Reproduction traits; Sperm DNA methylation; Variably methylated regions.
Conflict of interest statement
The authors declare that they have no competing interests except that George Liu is a member of the editorial board (Associate Editor) of this journal.
Figures
References
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
