

The Hyaluronidase, TMEM2, Promotes ER Homeostasis and Longevity Independent of the UPRER
- PMID: 31761535
- PMCID: PMC6913896
- DOI: 10.1016/j.cell.2019.10.018
The Hyaluronidase, TMEM2, Promotes ER Homeostasis and Longevity Independent of the UPRER
Abstract
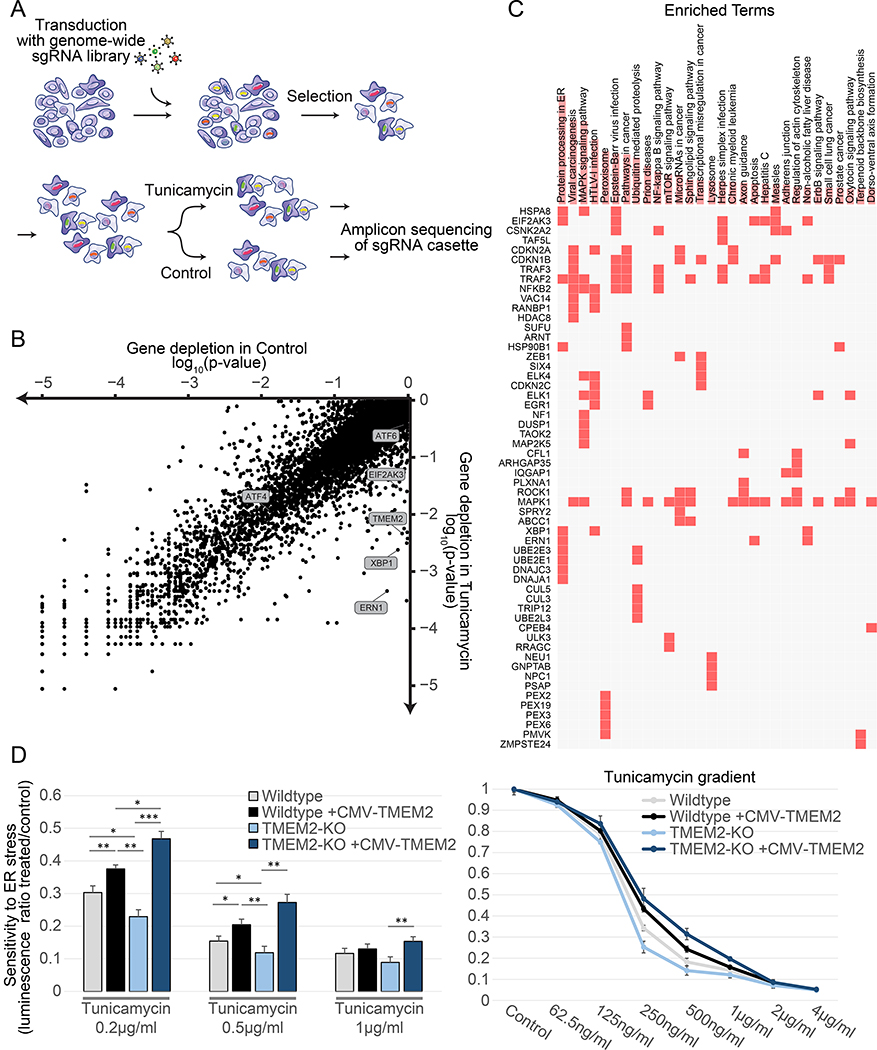
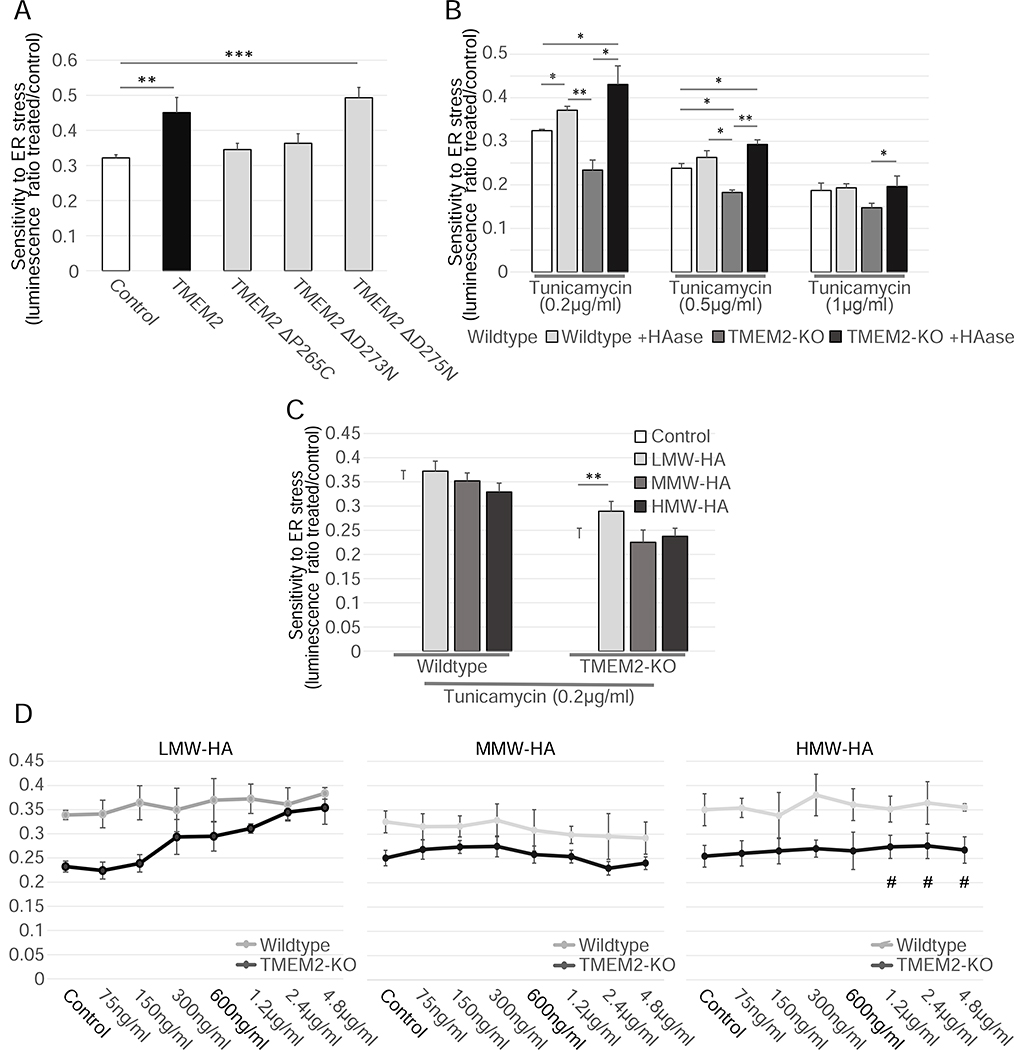
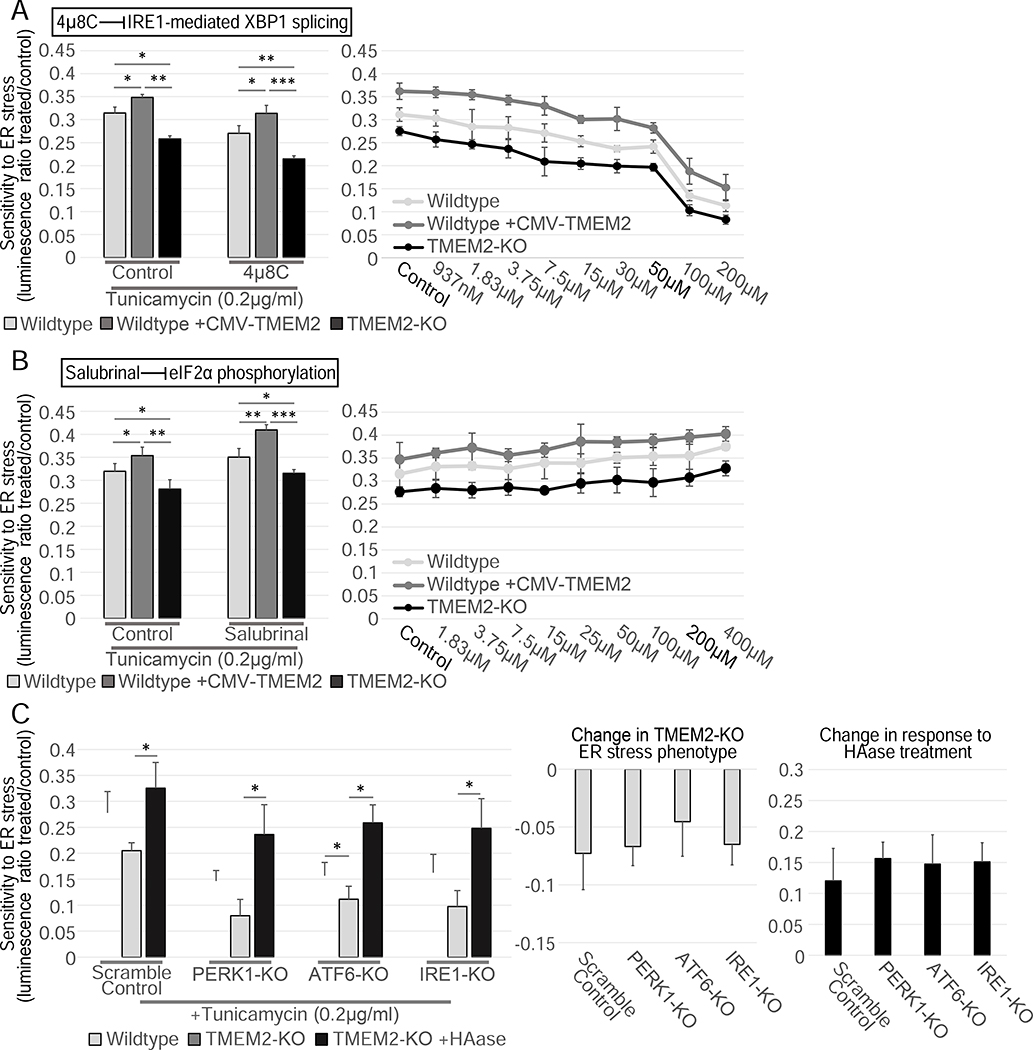
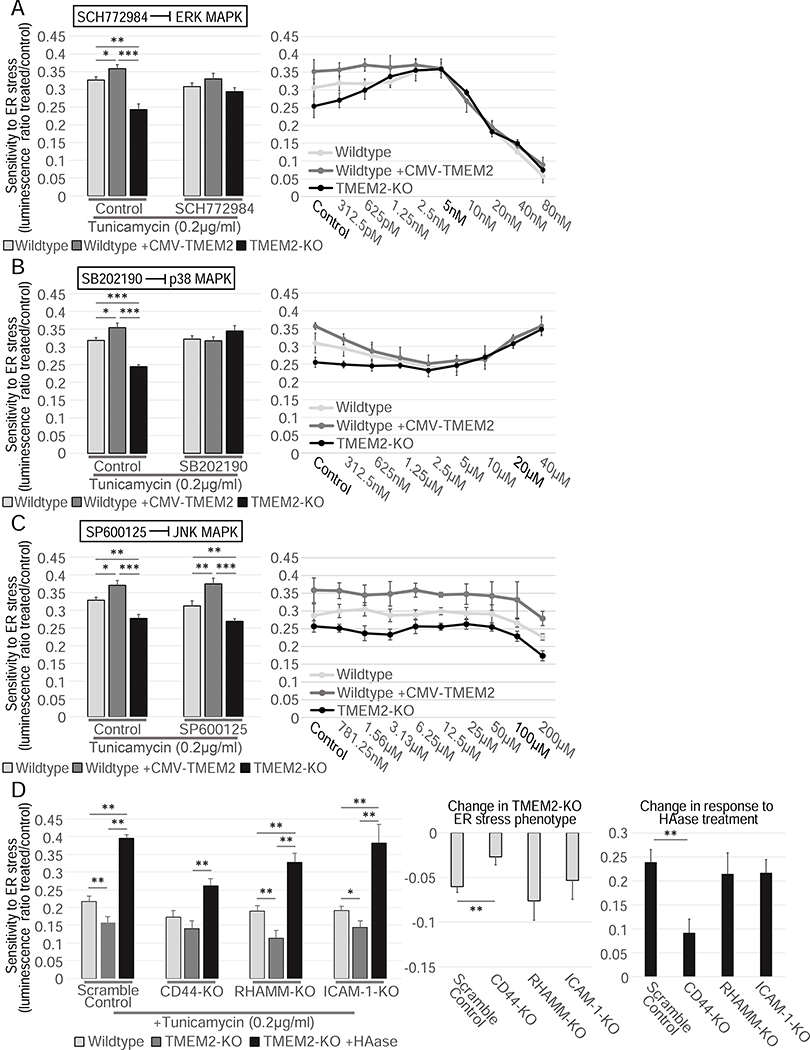
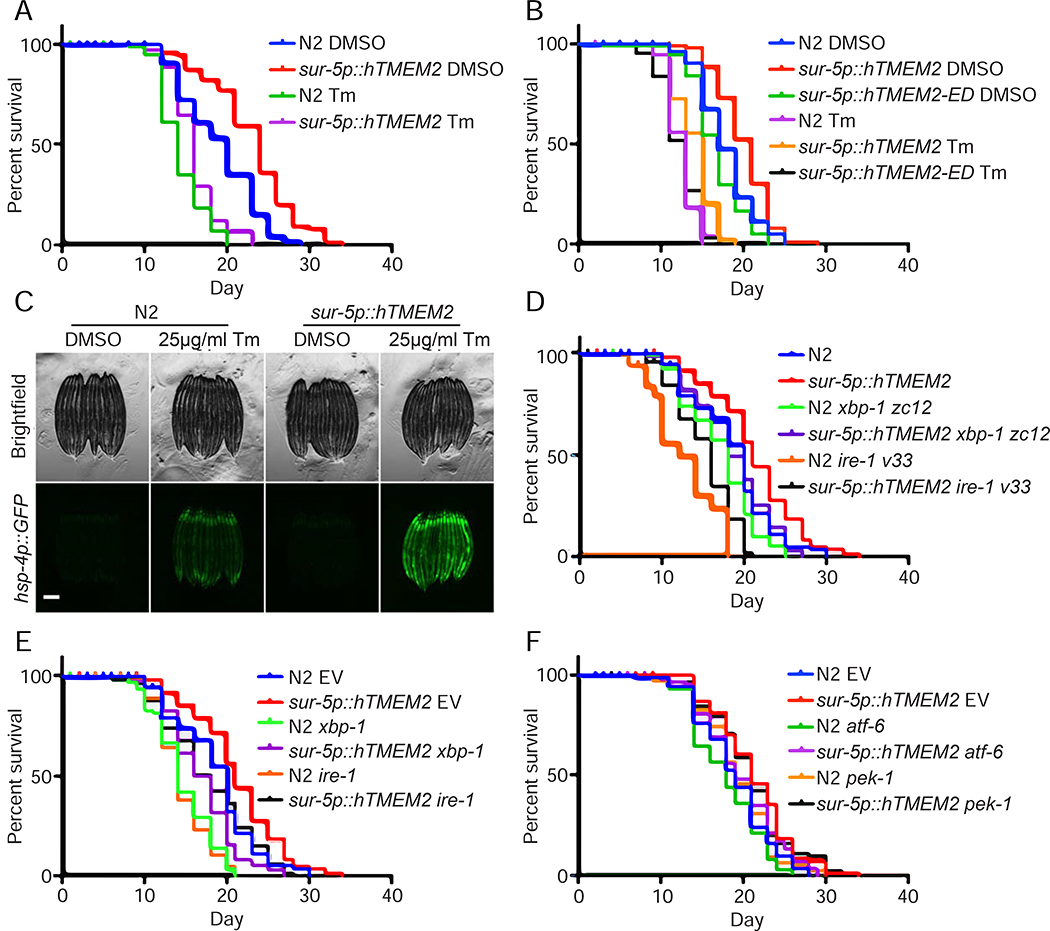
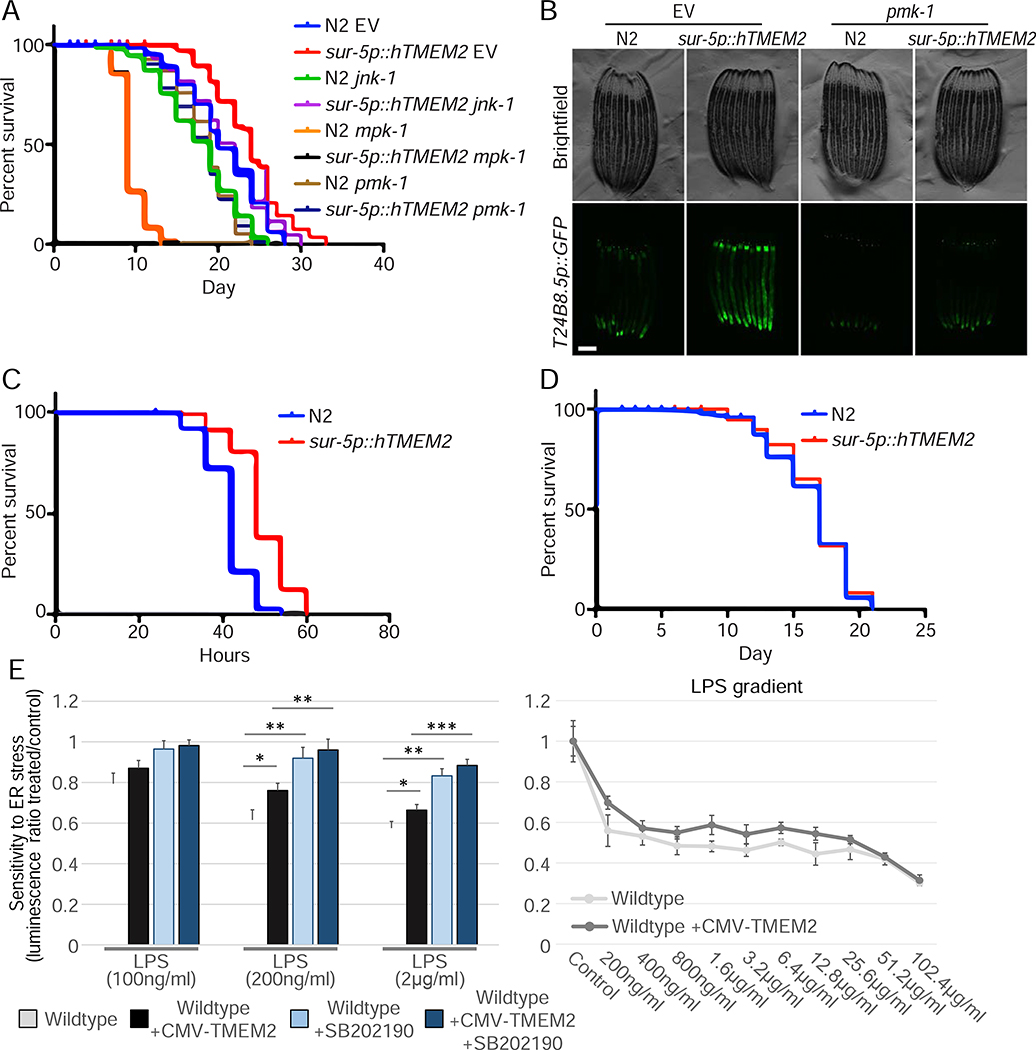
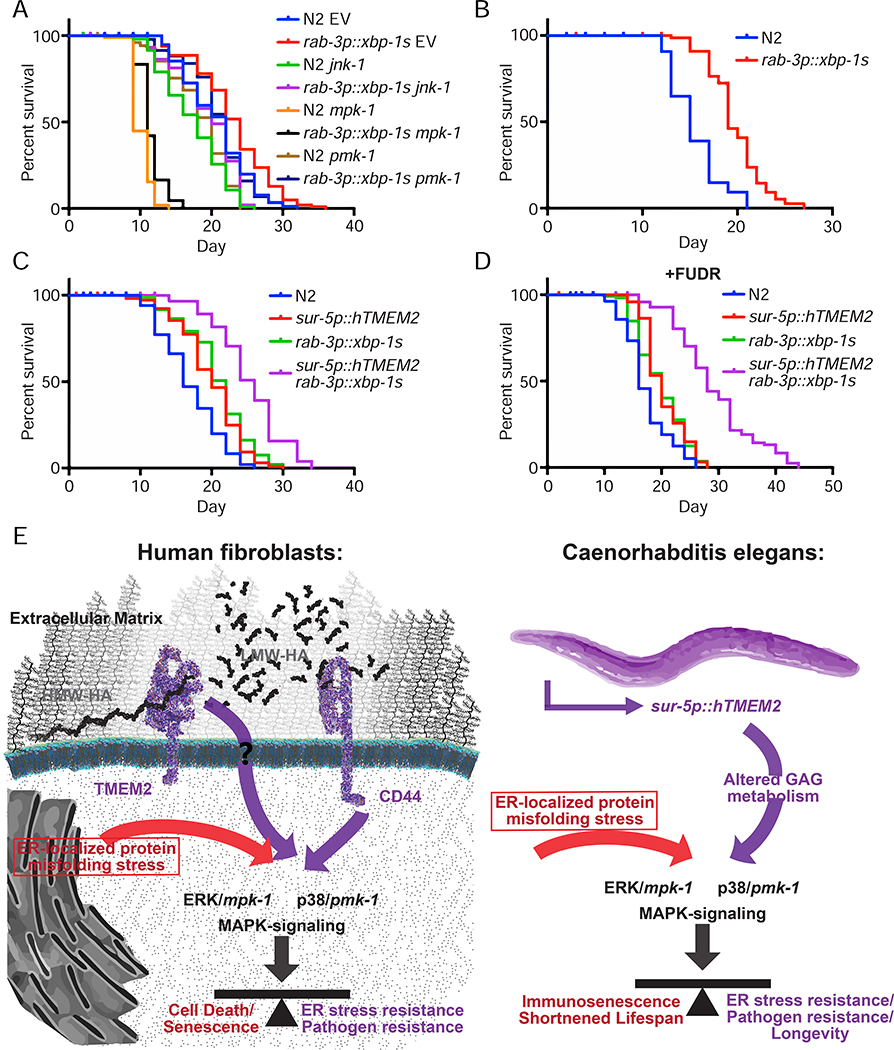
Cells have evolved complex mechanisms to maintain protein homeostasis, such as the UPRER, which are strongly associated with several diseases and the aging process. We performed a whole-genome CRISPR-based knockout (KO) screen to identify genes important for cells to survive ER-based protein misfolding stress. We identified the cell-surface hyaluronidase (HAase), Transmembrane Protein 2 (TMEM2), as a potent modulator of ER stress resistance. The breakdown of the glycosaminoglycan, hyaluronan (HA), by TMEM2 within the extracellular matrix (ECM) altered ER stress resistance independent of canonical UPRER pathways but dependent upon the cell-surface receptor, CD44, a putative HA receptor, and the MAPK cell-signaling components, ERK and p38. Last, and most surprisingly, ectopic expression of human TMEM2 in C. elegans protected animals from ER stress and increased both longevity and pathogen resistance independent of canonical UPRER activation but dependent on the ERK ortholog mpk-1 and the p38 ortholog pmk-1.
Keywords: CRISPR-Cas9; MAPK signaling; aging; endoplasmic reticulum; extracellular matrix; glucosaminoglycan; immune response; stress response.
Copyright © 2019 Elsevier Inc. All rights reserved.
Conflict of interest statement
Competing Financial Interests/Declaration of Interests
The authors declare no competing interests.
Figures
Comment in
-
Saved by the Matrix: UPR Independent Survival under ER Stress.Cell. 2019 Nov 27;179(6):1246-1248. doi: 10.1016/j.cell.2019.11.012. Cell. 2019. PMID: 31778650
-
TMEM2 Modulates ER Stress in a Non-canonical Manner.Cell Metab. 2019 Dec 3;30(6):999-1001. doi: 10.1016/j.cmet.2019.11.008. Cell Metab. 2019. PMID: 31801059
References
-
- Adamson B, Norman TM, Jost M, Cho MY, Nuñez JK, Chen Y, Villalta JE, Gilbert LA, Horlbeck MA, Hein MY, Pak RA, Gray AN, Gross CA, Dixit A, Parnas O, Regev A, Weissman JS, 2016. A Multiplexed Single-Cell CRISPR Screening Platform Enables Systematic Dissection of the Unfolded Protein Response. Cell 167, 1867–1882.e21. 10.1016/j.cell.2016.11.048 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
