

A Novel SLC1A4 Mutation (p.Y191*) Causes Spastic Tetraplegia, Thin Corpus Callosum, and Progressive Microcephaly (SPATCCM) With Seizure Disorder
- PMID: 31763347
- PMCID: PMC6852354
- DOI: 10.1177/2329048X19880647
A Novel SLC1A4 Mutation (p.Y191*) Causes Spastic Tetraplegia, Thin Corpus Callosum, and Progressive Microcephaly (SPATCCM) With Seizure Disorder
Abstract
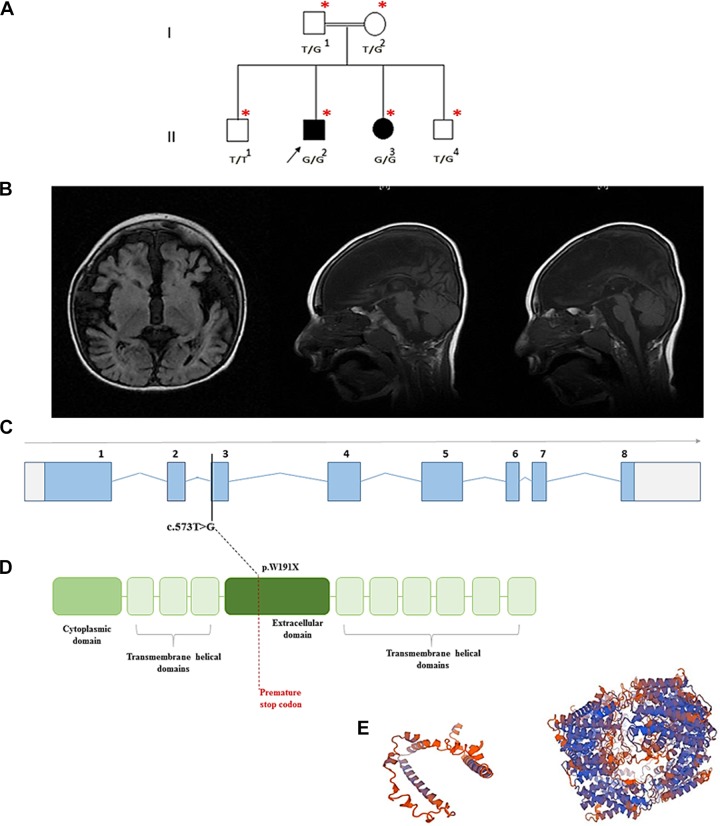
Spastic tetraplegia, thin corpus callosum, and progressive microcephaly is a recently described very rare autosomal recessive neurodevelopmental disorder. This disease was first described in 2015 in several families from the Ashkenazi Jewish ancestry with a founder mutation in SLC1A4 (p.E256K) as the underlying genetic cause. SLC1A4 gene encodes for the amino acid transporter ASCT1 that is necessary for serine cellular transport to neurons. We clinically evaluated 2 Pakistani siblings with severe global developmental delay, progressive microcephaly, and seizure disorder. We performed exome sequencing, Sanger sequencing, and segregation analysis to identify the genetic cause of the phenotype followed by in silico analysis to evaluate the pathogenicity of the identified mutation. We identified a novel homozygous variant (c.573T>G) in both patients. The mutation is predicted to cause nonsense mutation (p.Y191*) in the ASCT1 protein. Here, we report the fifth disease causing mutation in SLC1A4 gene and review all previously reported cases.
Keywords: developmental delay; epileptic encephalopathy; infantile spasms; next-generation sequencing; spasticity.
© The Author(s) 2019.
Conflict of interest statement
Declaration of Conflicting Interests: The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Figures
References
-
- Damseh N, Simonin A, Jalas C, et al. Mutations in SLC1A4, encoding the brain serine transporter, are associated with developmental delay, microcephaly and hypomyelination. J Med Genet. 2015;52(8):541–547. doi:10.1136/jmedgenet-2015-103104. - PubMed
-
- Heimer G, Marek-Yagel D, Eyal E, et al. SLC1A4 mutations cause a novel disorder of intellectual disability, progressive microcephaly, spasticity and thin corpus callosum. Clin Genet. 2015;88(4):327–335. doi:10.1111/cge.12637. - PubMed
-
- Srour M, Hamdan FF, Gan-Or Z, et al. A homozygous mutation in SLC1A4 in siblings with severe intellectual disability and microcephaly. Clin Genet. 2015;88:E1–E4. doi:10.1111/cge.12605. - PubMed
-
- Conroy J, Allen NM, Gorman K, et al. Novel European SLC1A4 variant: infantile spasms and population ancestry analysis. J Hum Genet. 2016;61(8):761–764. doi:10.1038/jhg.2016.44. - PubMed
-
- Kent WJ, Sugnet CW, Furey TS, Roskin KM. The human genome browser at UCSC W. J Med Chem. 1976;19(10):1228–1231. doi:10.1101/gr.229102.
LinkOut - more resources
Full Text Sources
