

Stalled developmental programs at the root of pediatric brain tumors
- PMID: 31768071
- PMCID: PMC6885128
- DOI: 10.1038/s41588-019-0531-7
Stalled developmental programs at the root of pediatric brain tumors
Abstract
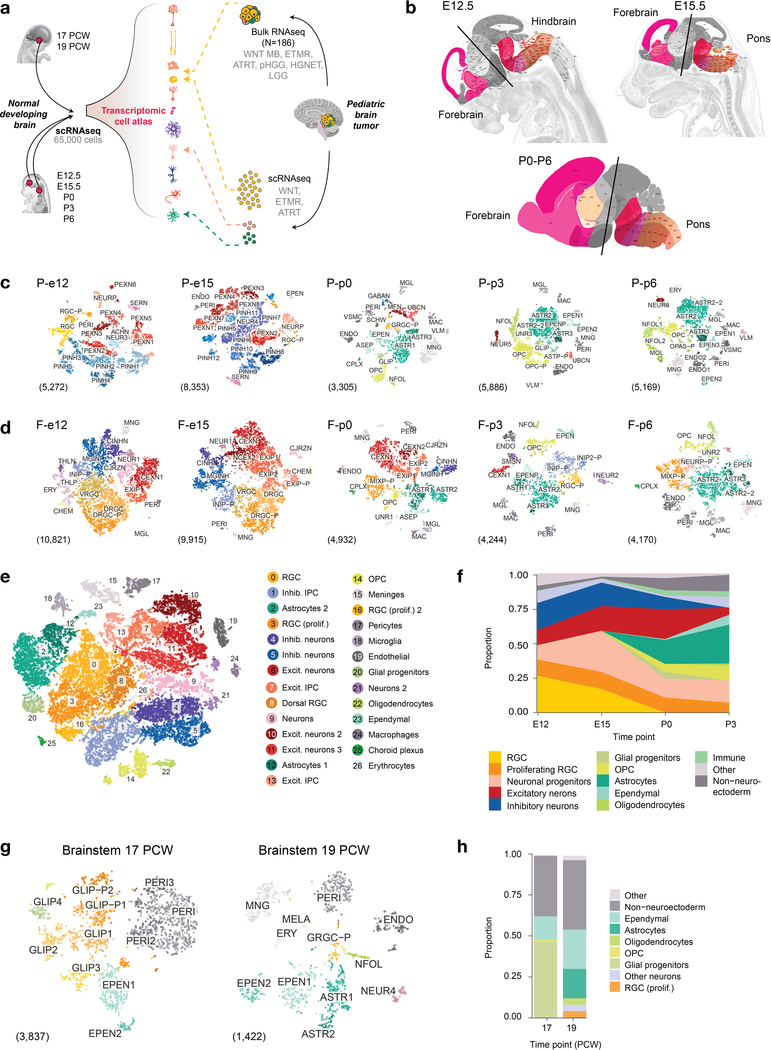
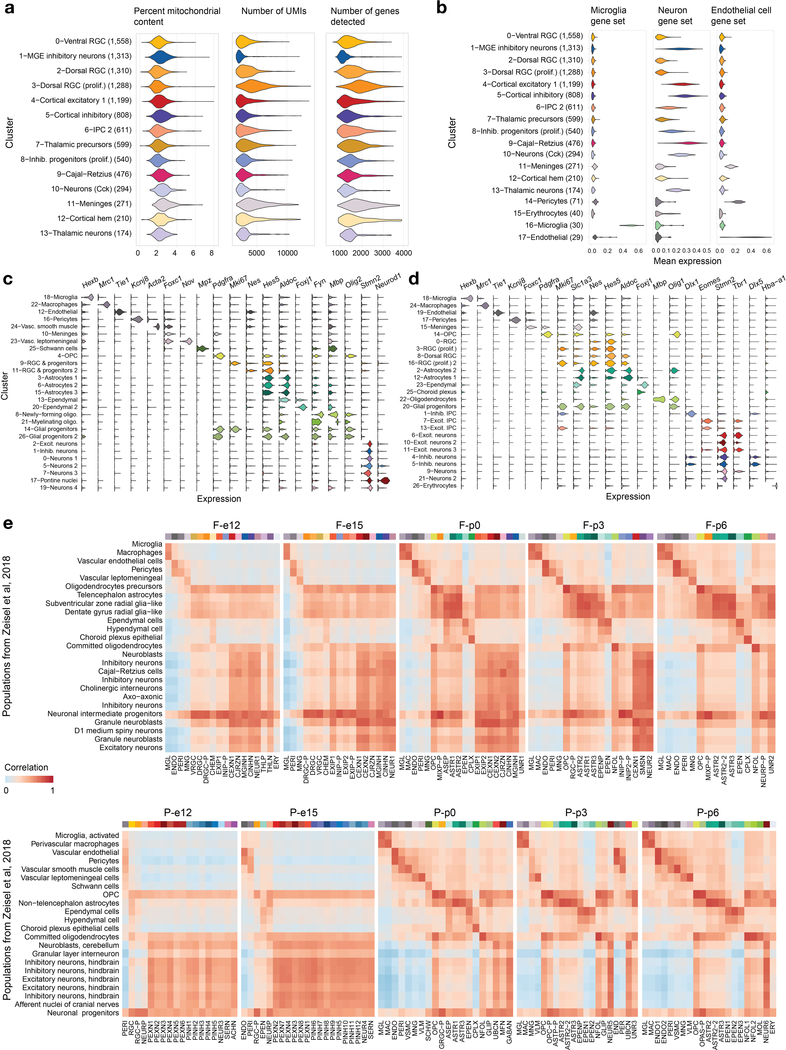
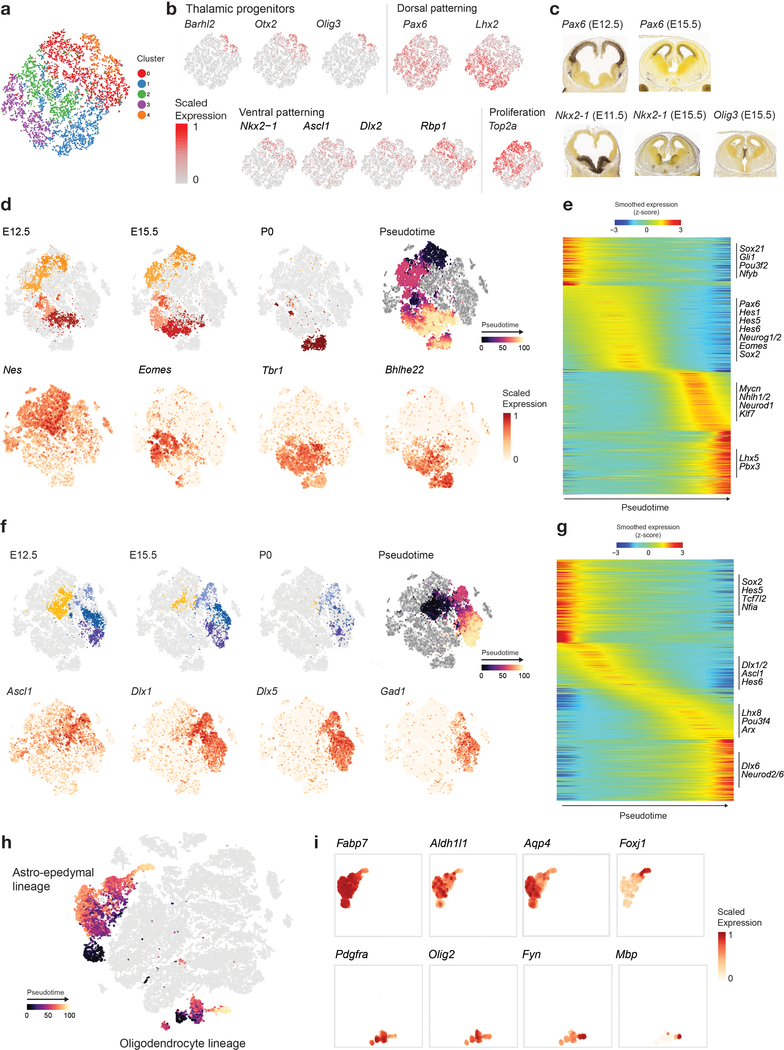
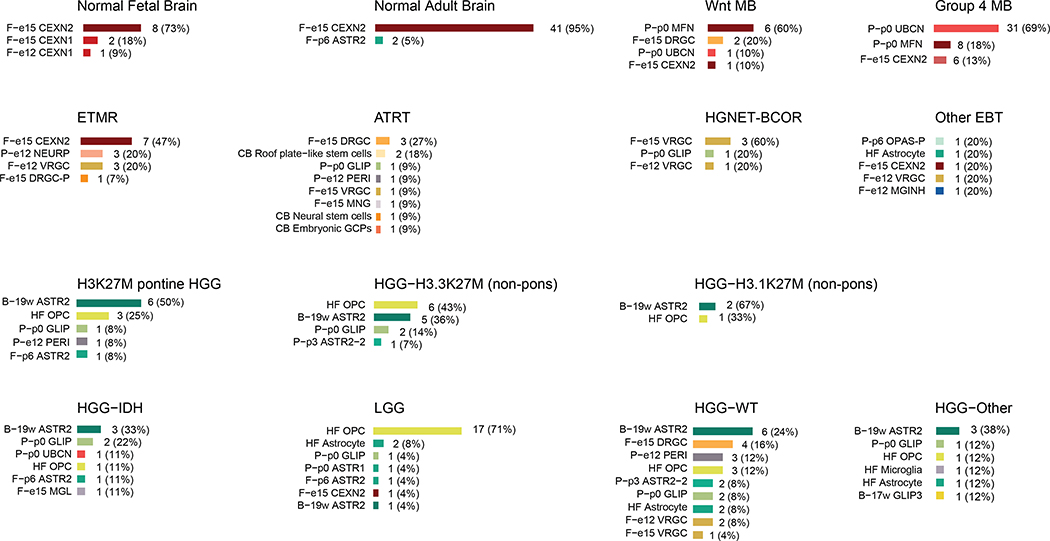
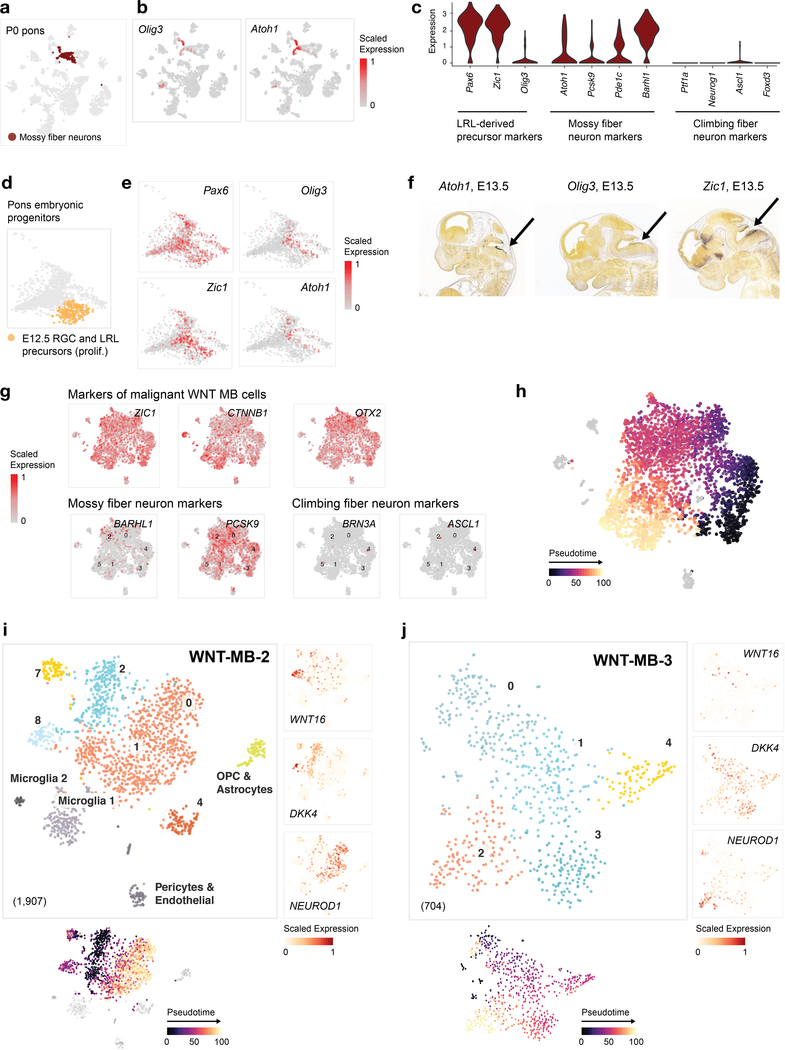
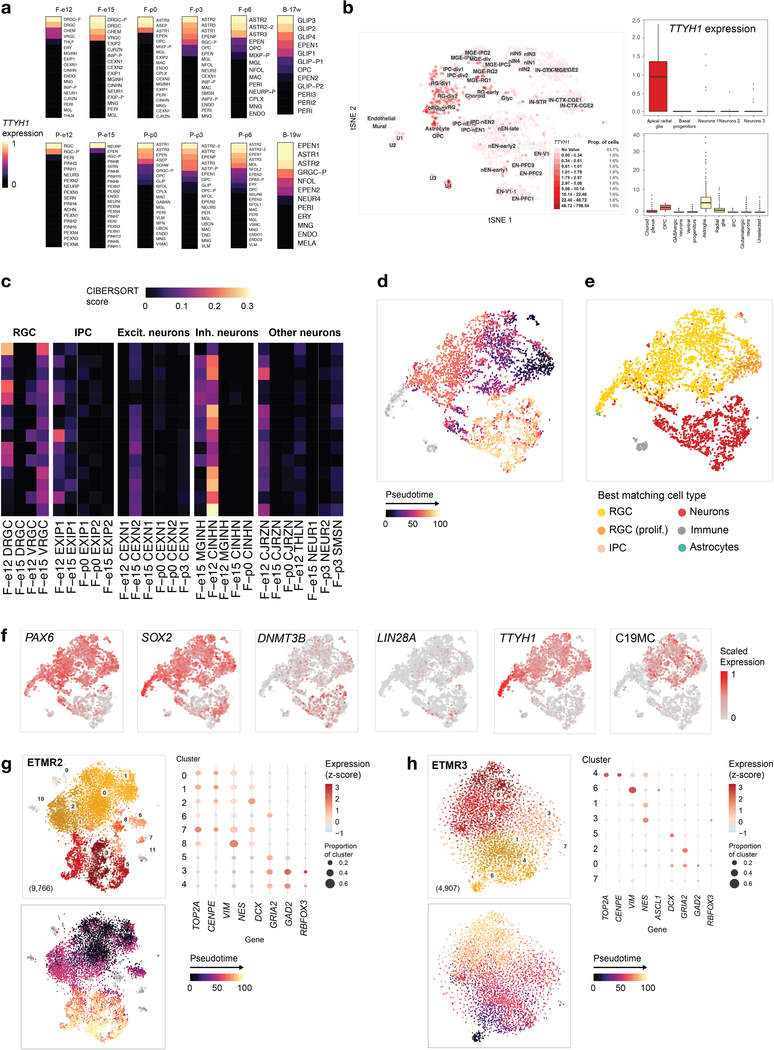
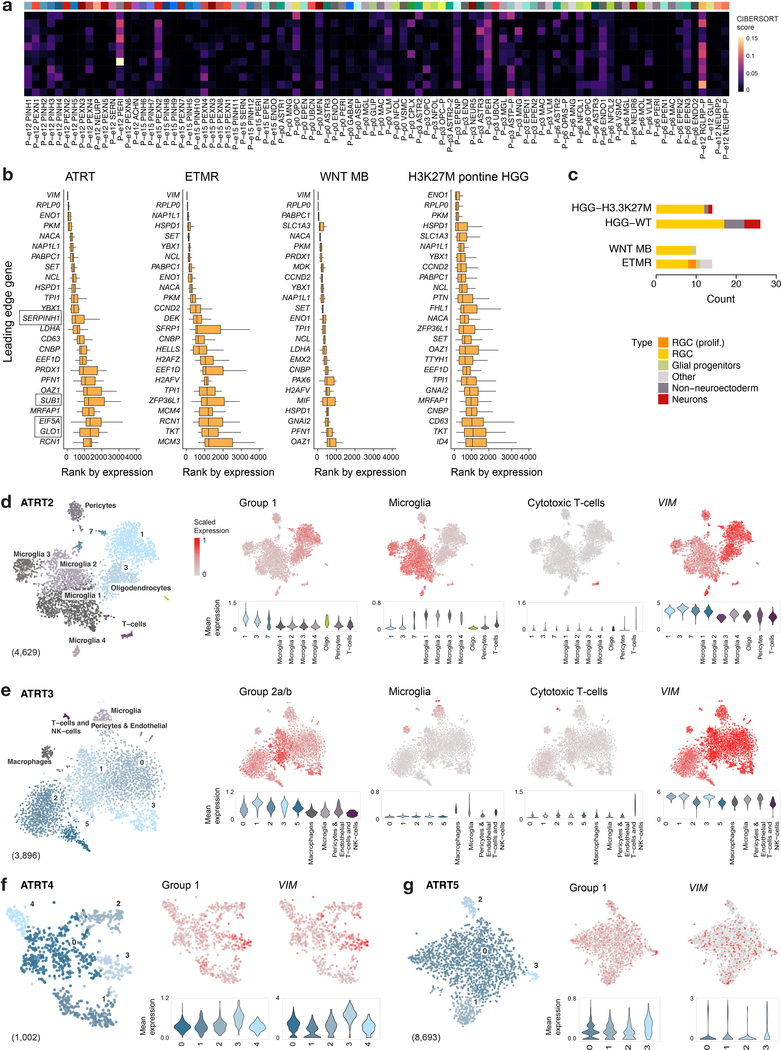
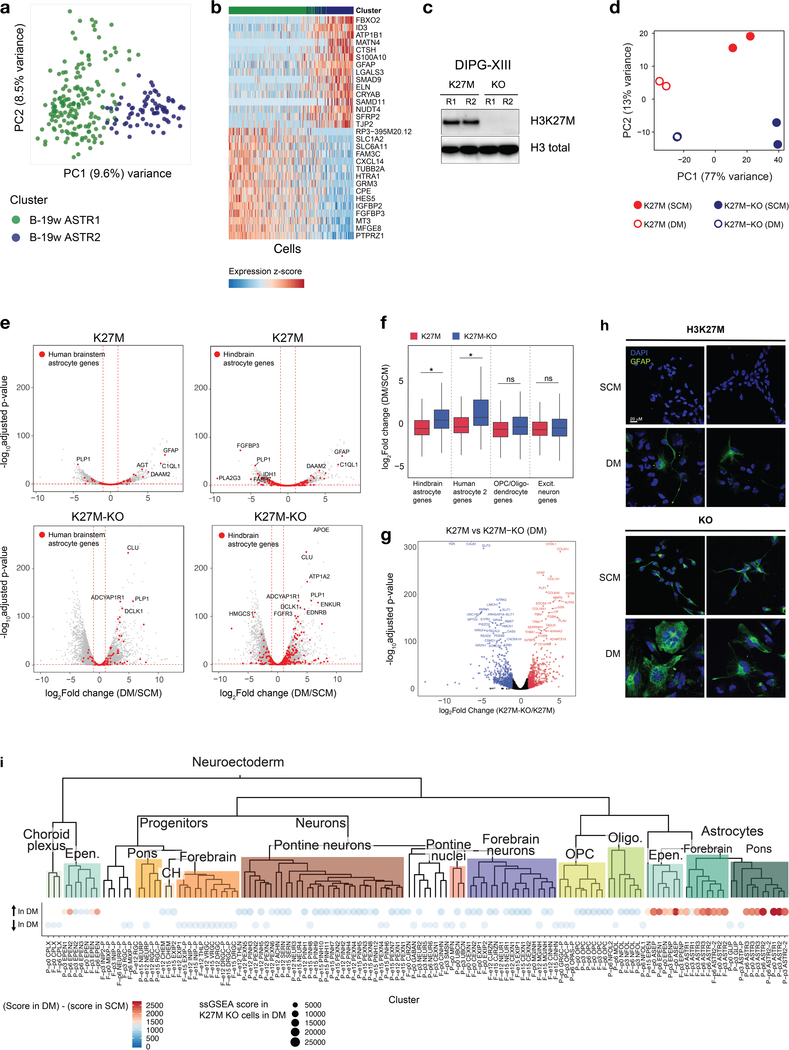
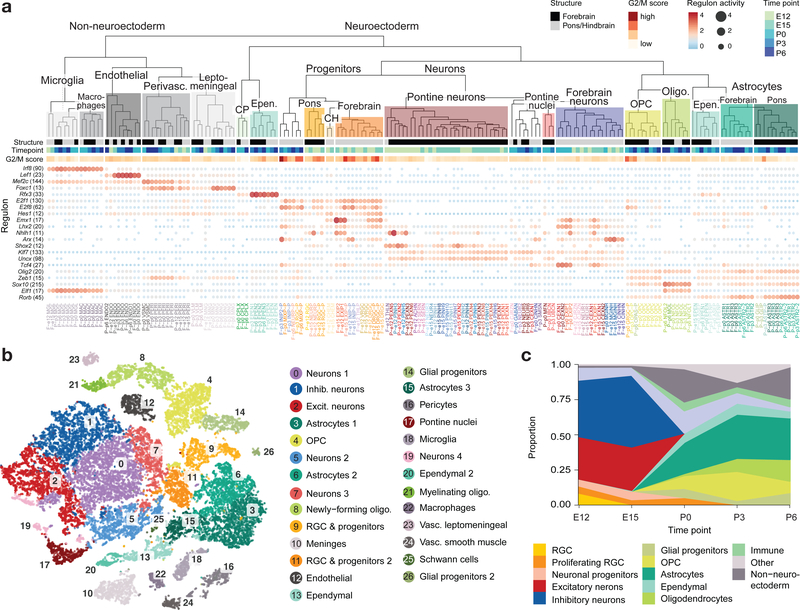
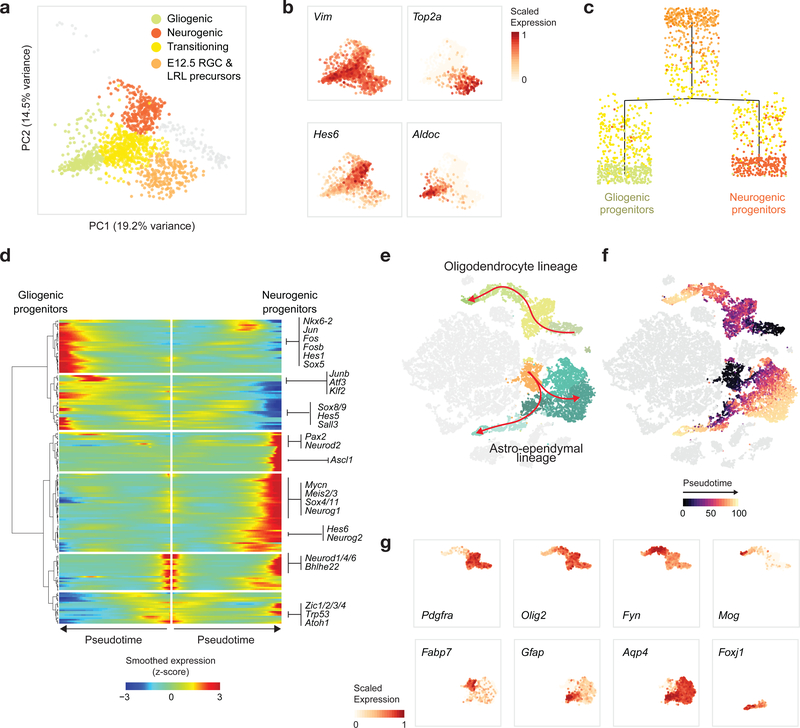
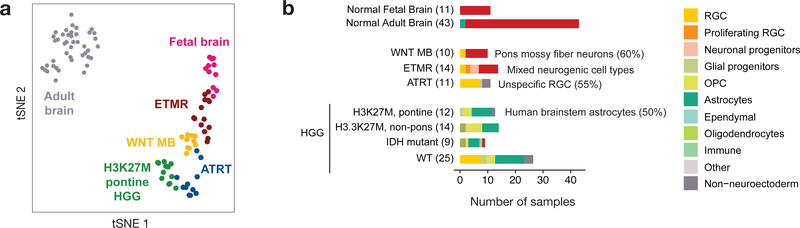
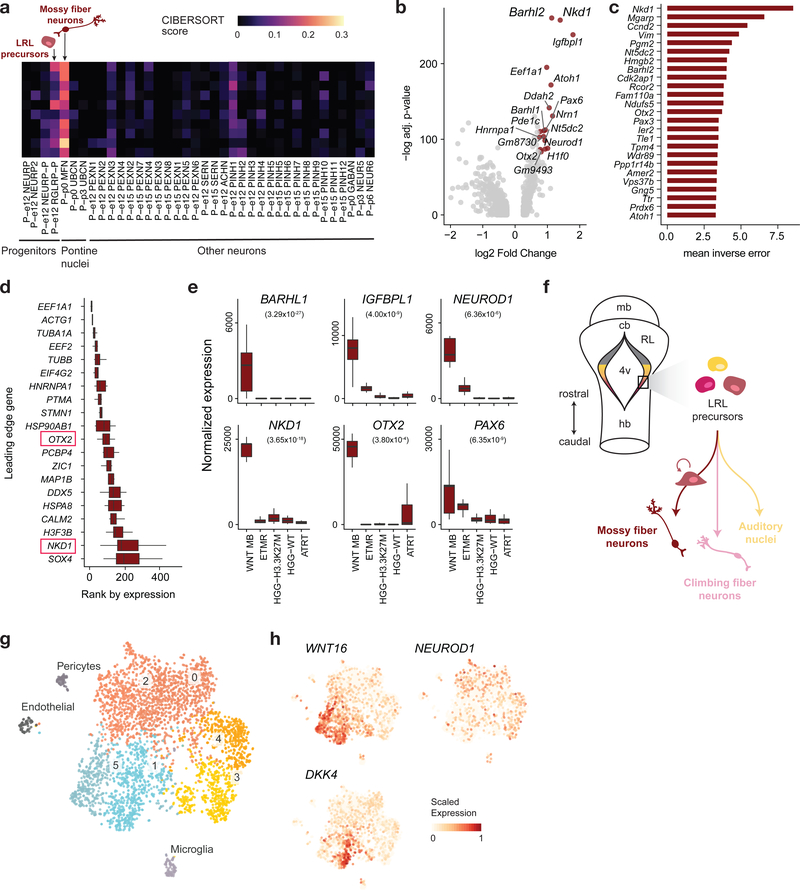
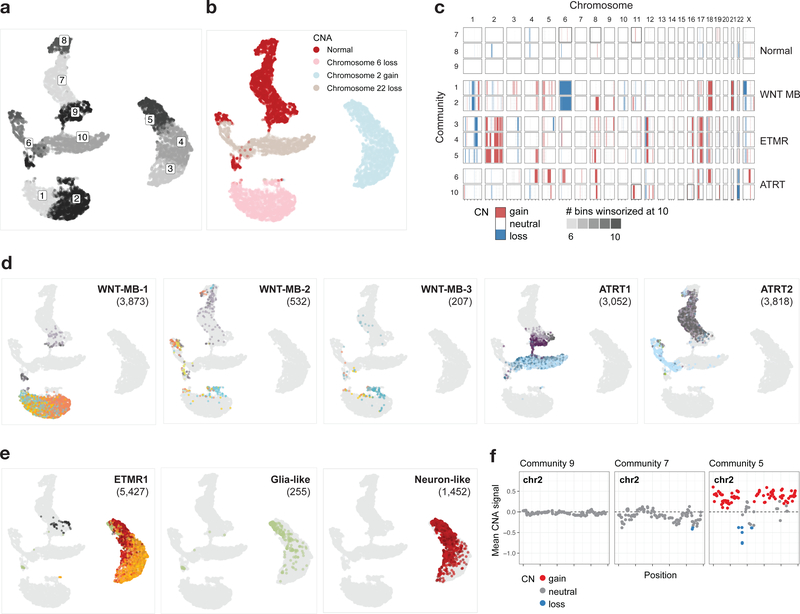
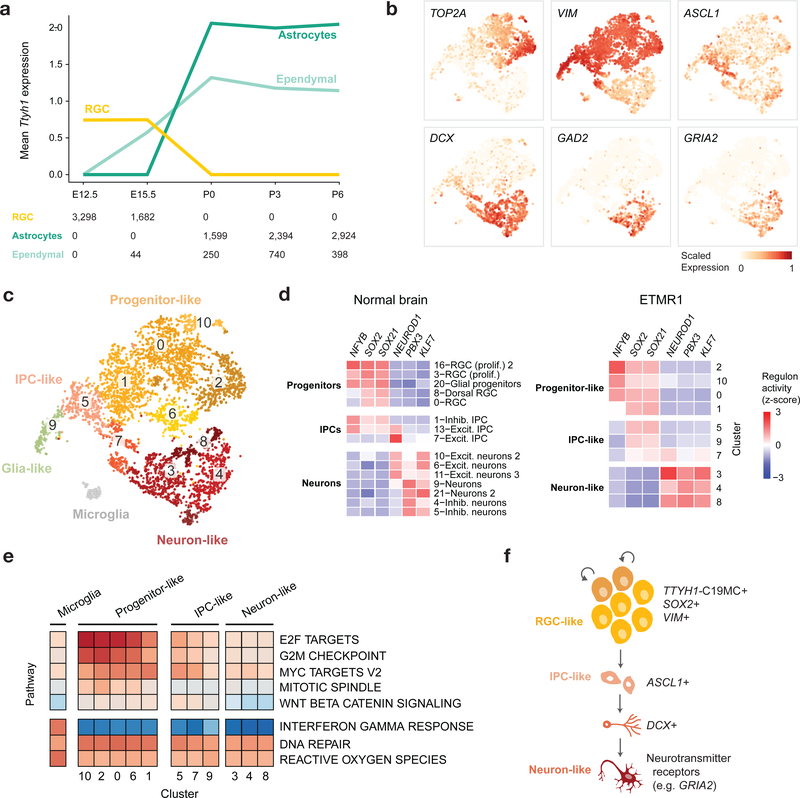
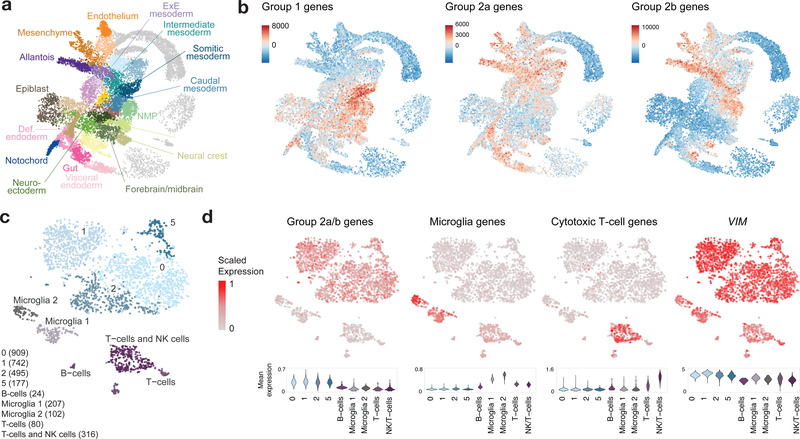
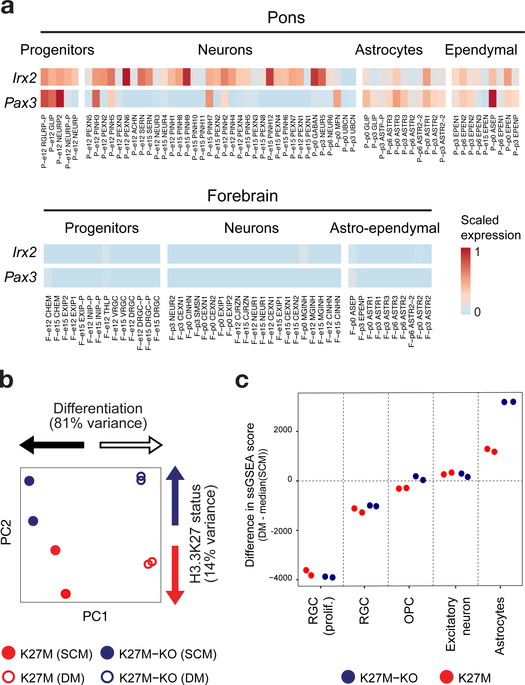
Childhood brain tumors have suspected prenatal origins. To identify vulnerable developmental states, we generated a single-cell transcriptome atlas of >65,000 cells from embryonal pons and forebrain, two major tumor locations. We derived signatures for 191 distinct cell populations and defined the regional cellular diversity and differentiation dynamics. Projection of bulk tumor transcriptomes onto this dataset shows that WNT medulloblastomas match the rhombic lip-derived mossy fiber neuronal lineage and embryonal tumors with multilayered rosettes fully recapitulate a neuronal lineage, while group 2a/b atypical teratoid/rhabdoid tumors may originate outside the neuroectoderm. Importantly, single-cell tumor profiles reveal highly defined cell hierarchies that mirror transcriptional programs of the corresponding normal lineages. Our findings identify impaired differentiation of specific neural progenitors as a common mechanism underlying these pediatric cancers and provide a rational framework for future modeling and therapeutic interventions.
Conflict of interest statement
Competing Interests Statement
The authors declare no competing interests
Figures
References
-
- Kieran MW, Walker D, Frappaz D & Prados M Brain tumors: from childhood through adolescence into adulthood. J Clin Oncol 28, 4783–4789 (2010). - PubMed
-
- Jacob K et al. Genetic aberrations leading to MAPK pathway activation mediate oncogene-induced senescence in sporadic pilocytic astrocytomas. Clin. Cancer Res. 17, 4650–4660 (2011). - PubMed
-
- Johann PD et al. Atypical teratoid/rhabdoid tumors are comprised of three epigenetic subgroups with distinct enhancer landscapes. Cancer Cell 29, 379–393 (2016). - PubMed
Methods-only references
-
- Nagy C et al. Single-nucleus RNA sequencing shows convergent evidence from different cell types for altered synaptic plasticity in major depressive disorder. bioRxiv, 384479, doi: 10.1101/384479 (2019). - DOI
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
