

Microphysiological systems modeling acute respiratory distress syndrome that capture mechanical force-induced injury-inflammation-repair
- PMID: 31768486
- PMCID: PMC6874511
- DOI: 10.1063/1.5111549
Microphysiological systems modeling acute respiratory distress syndrome that capture mechanical force-induced injury-inflammation-repair
Abstract
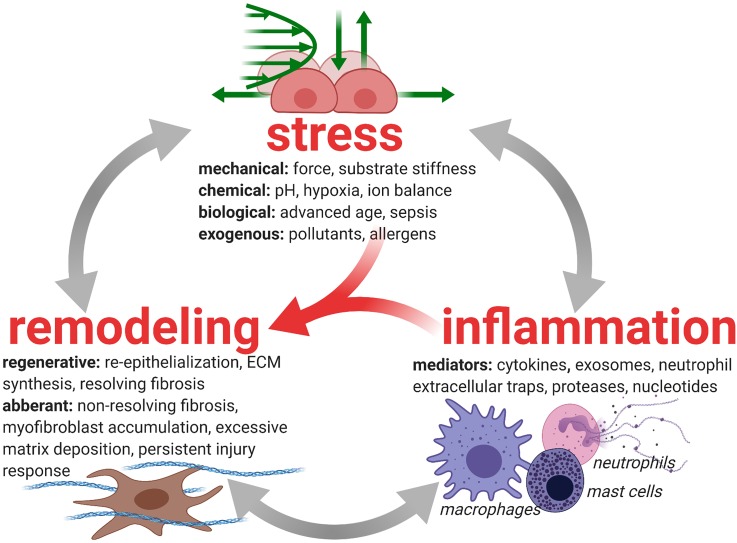
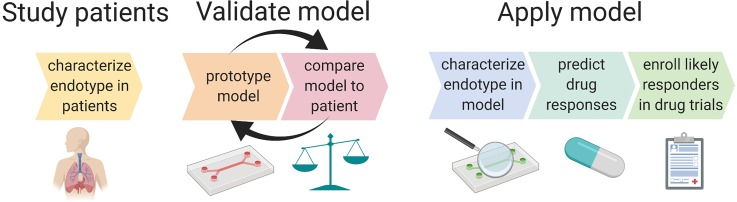
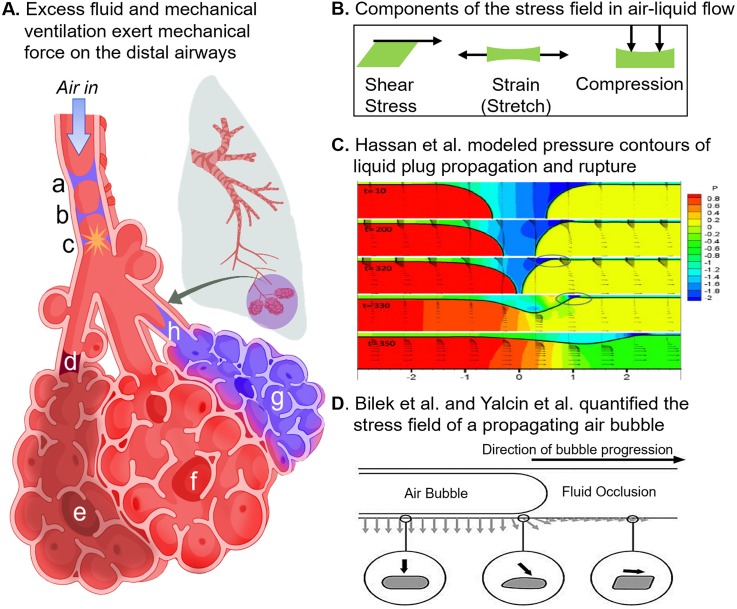
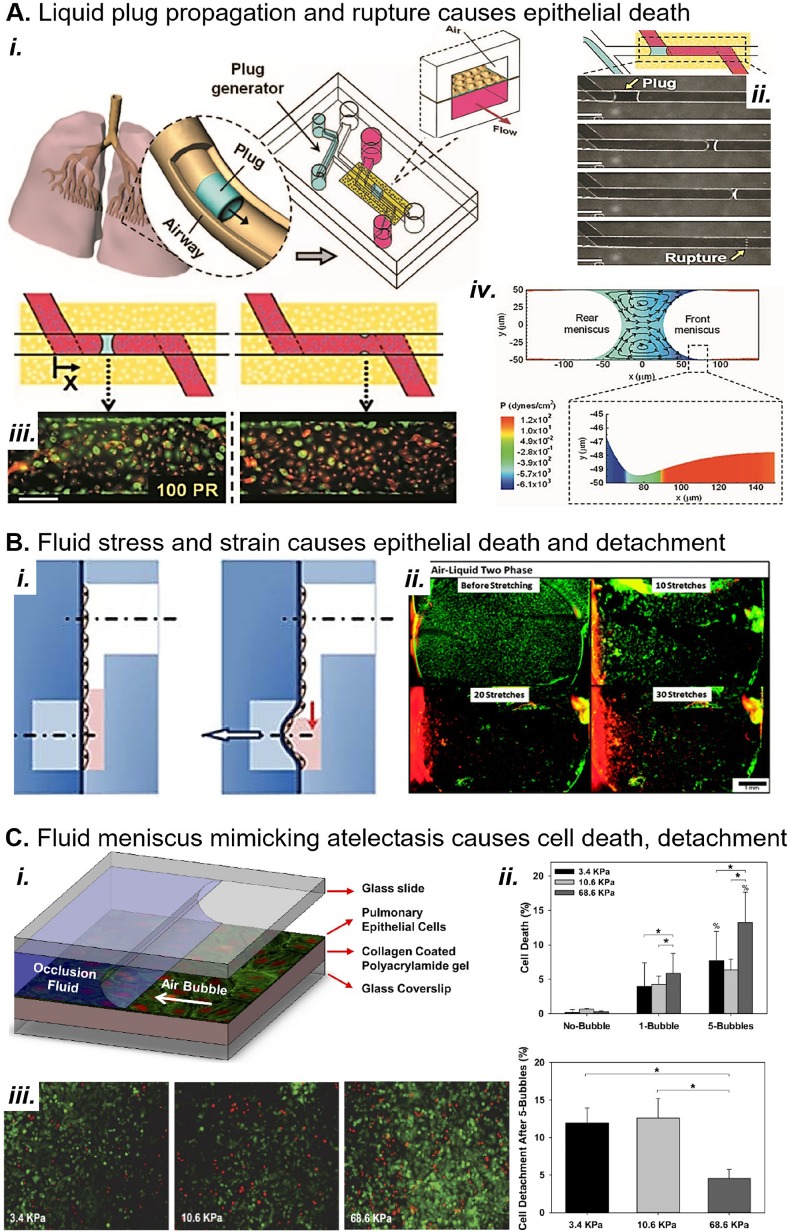
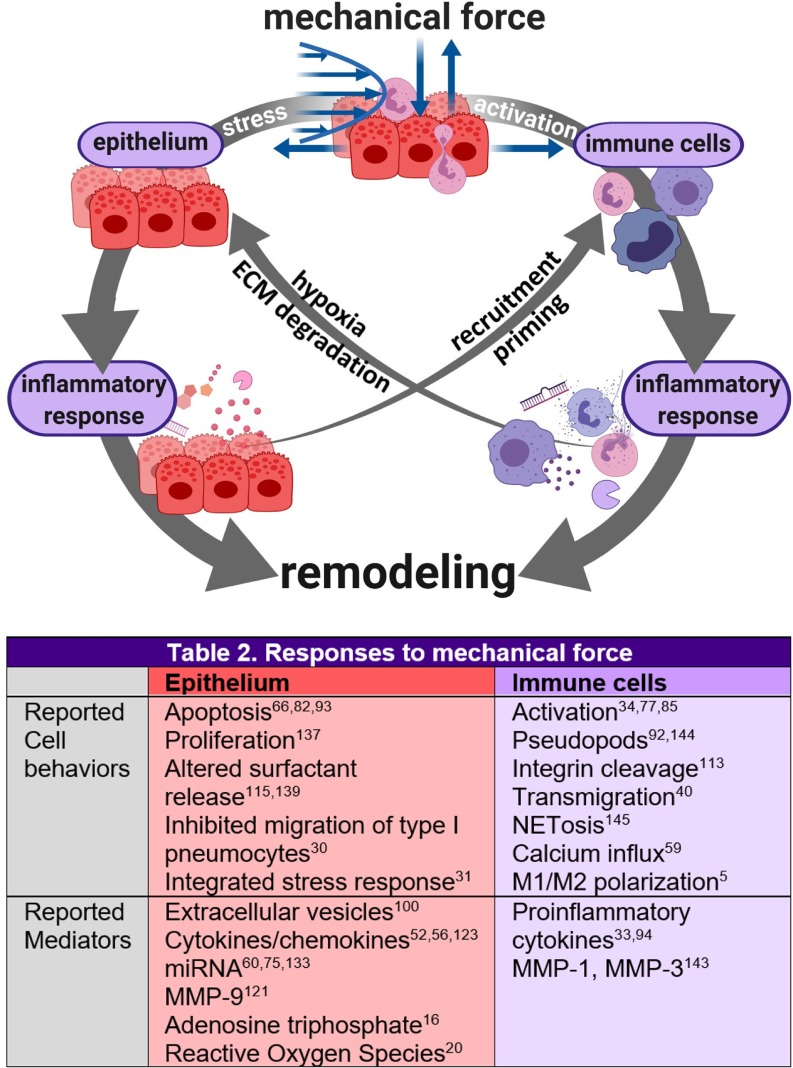
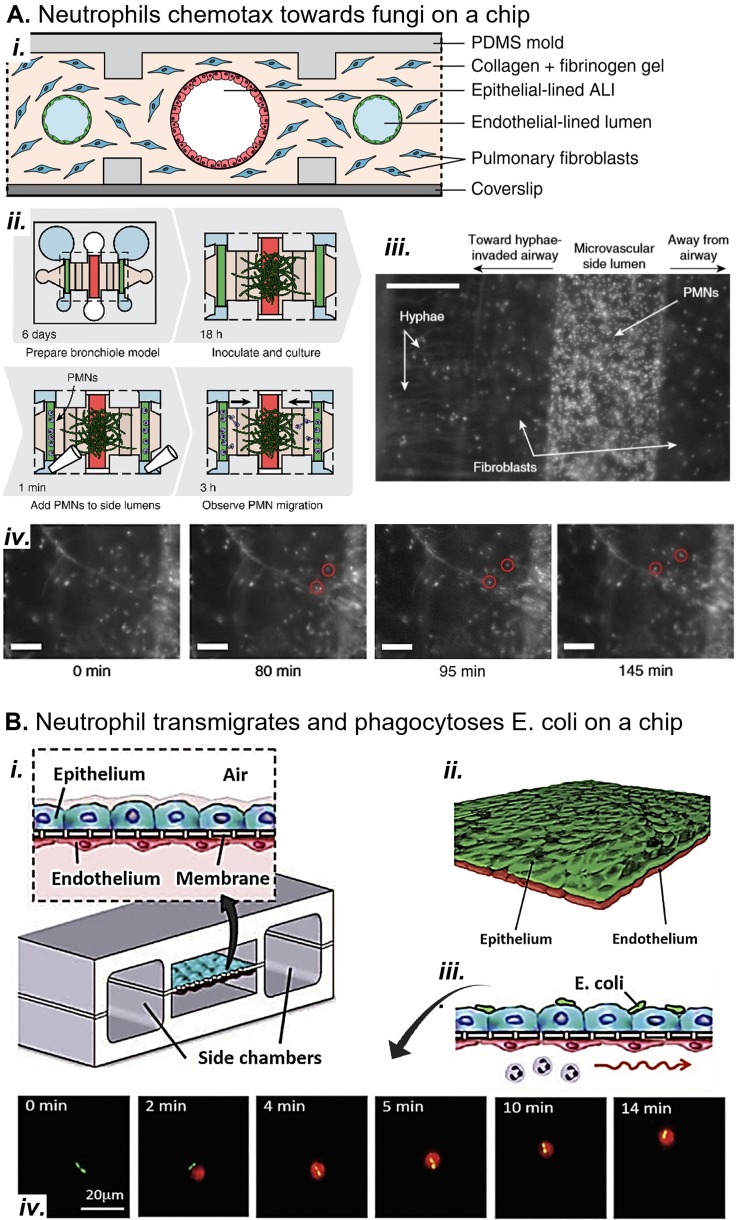
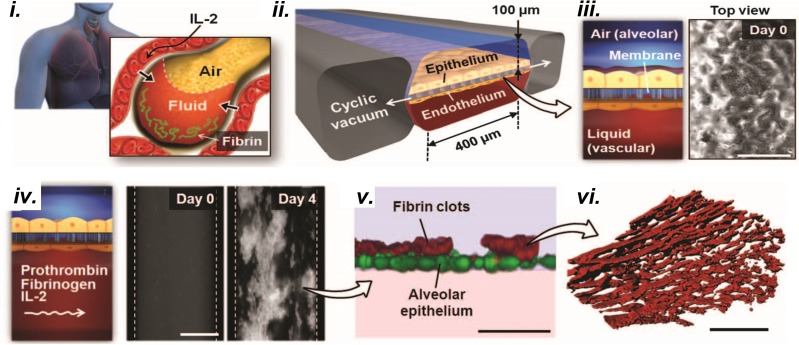
Complex in vitro models of the tissue microenvironment, termed microphysiological systems, have enormous potential to transform the process of discovering drugs and disease mechanisms. Such a paradigm shift is urgently needed in acute respiratory distress syndrome (ARDS), an acute lung condition with no successful therapies and a 40% mortality rate. Here, we consider how microphysiological systems could improve understanding of biological mechanisms driving ARDS and ultimately improve the success of therapies in clinical trials. We first discuss how microphysiological systems could explain the biological mechanisms underlying the segregation of ARDS patients into two clinically distinct phenotypes. Then, we contend that ARDS-mimetic microphysiological systems should recapitulate three critical aspects of the distal airway microenvironment, namely, mechanical force, inflammation, and fibrosis, and we review models that incorporate each of these aspects. Finally, we recognize the substantial challenges associated with combining inflammation, fibrosis, and/or mechanical force in microphysiological systems. Nevertheless, complex in vitro models are a novel paradigm for studying ARDS, and they could ultimately improve patient care.
© Author(s).
Figures
References
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources