

Recurrent arginine substitutions in the ACTG2 gene are the primary driver of disease burden and severity in visceral myopathy
- PMID: 31769566
- PMCID: PMC7720429
- DOI: 10.1002/humu.23960
Recurrent arginine substitutions in the ACTG2 gene are the primary driver of disease burden and severity in visceral myopathy
Abstract
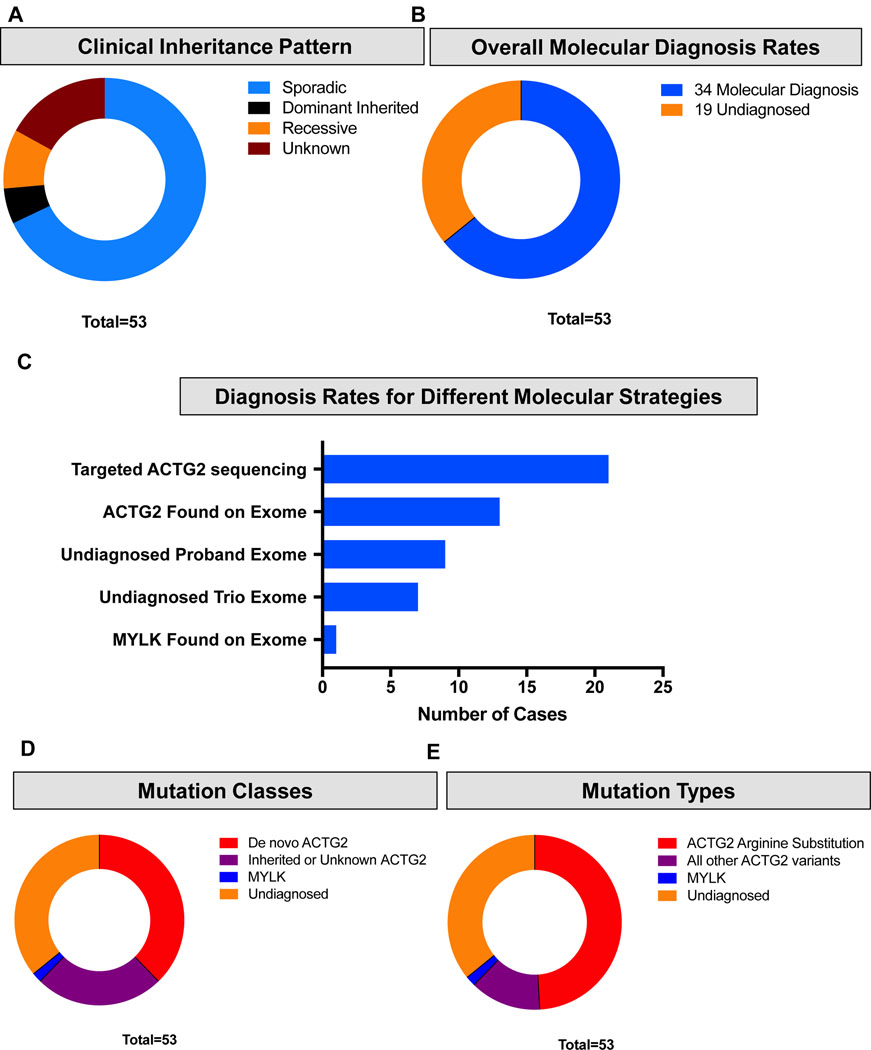
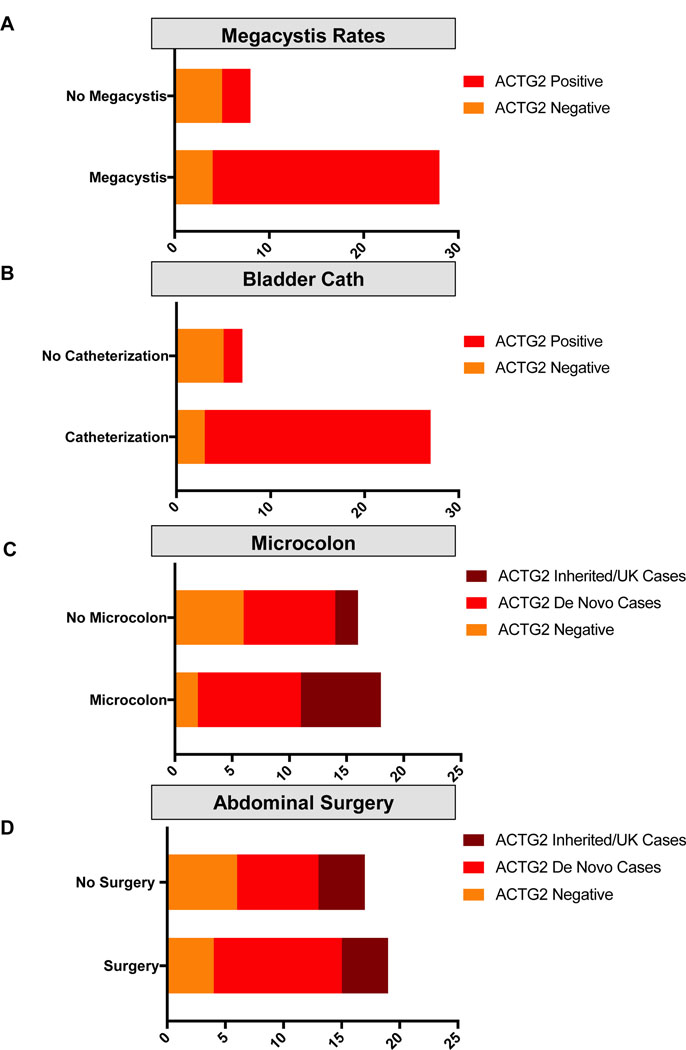
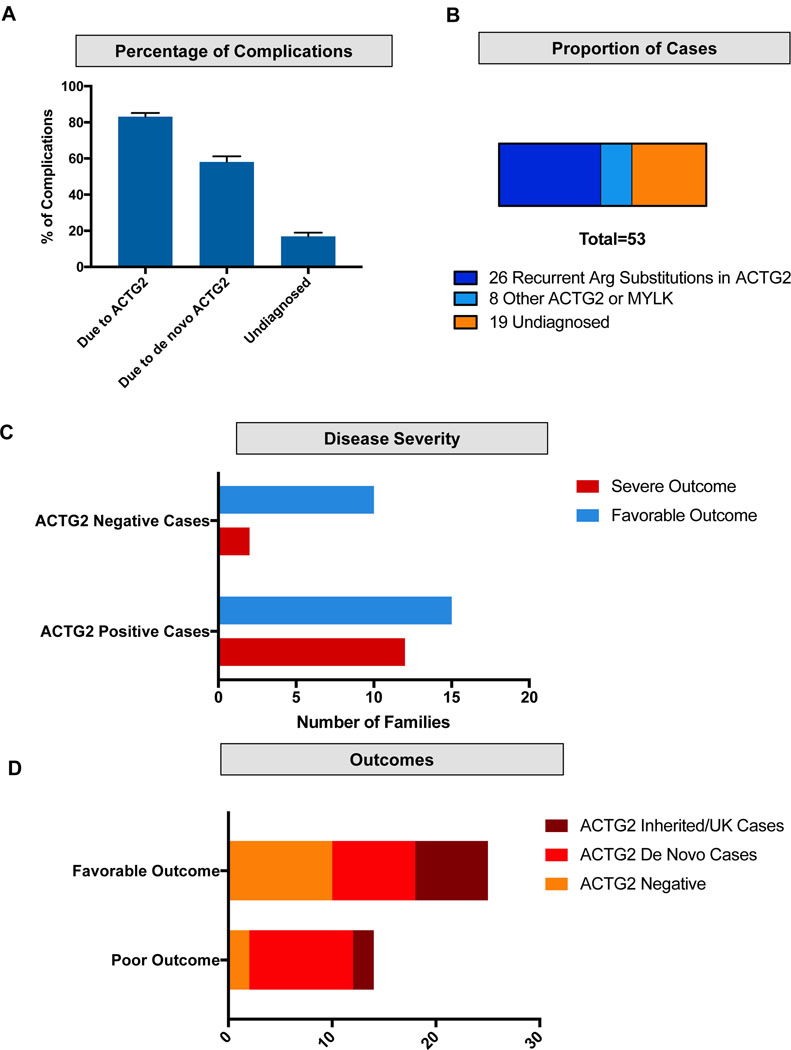
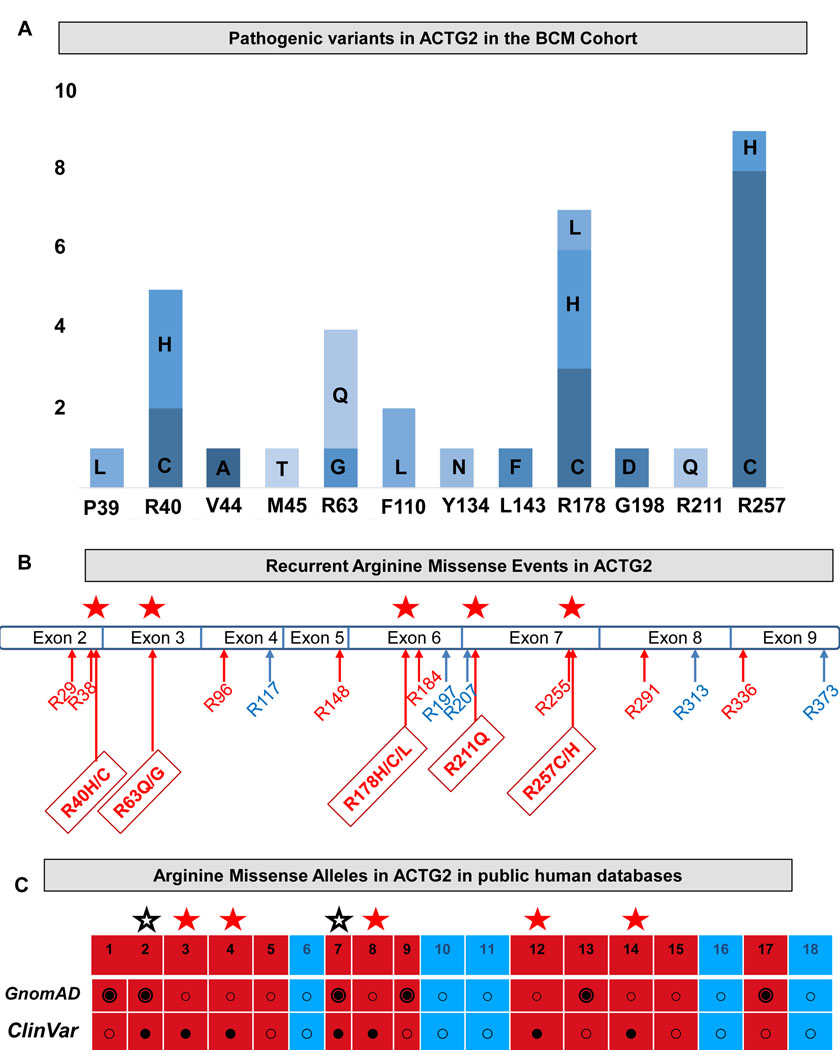
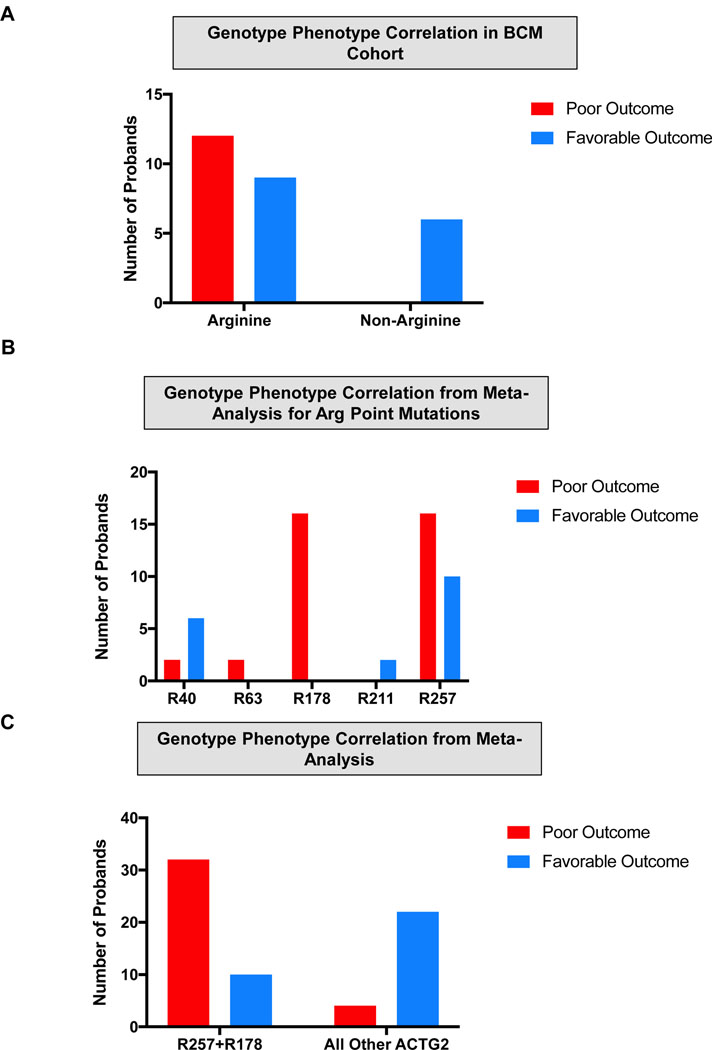
Visceral myopathy with abnormal intestinal and bladder peristalsis includes a clinical spectrum with megacystis-microcolon intestinal hypoperistalsis syndrome and chronic intestinal pseudo-obstruction. The vast majority of cases are caused by dominant variants in ACTG2; however, the overall genetic architecture of visceral myopathy has not been well-characterized. We ascertained 53 families, with visceral myopathy based on megacystis, functional bladder/gastrointestinal obstruction, or microcolon. A combination of targeted ACTG2 sequencing and exome sequencing was used. We report a molecular diagnostic rate of 64% (34/53), of which 97% (33/34) is attributed to ACTG2. Strikingly, missense mutations in five conserved arginine residues involving CpG dinucleotides accounted for 49% (26/53) of disease in the cohort. As a group, the ACTG2-negative cases had a more favorable clinical outcome and more restricted disease. Within the ACTG2-positive group, poor outcomes (characterized by total parenteral nutrition dependence, death, or transplantation) were invariably due to one of the arginine missense alleles. Analysis of specific residues suggests a severity spectrum of p.Arg178>p.Arg257>p.Arg40 along with other less-frequently reported sites p.Arg63 and p.Arg211. These results provide genotype-phenotype correlation for ACTG2-related disease and demonstrate the importance of arginine missense changes in visceral myopathy.
Keywords: ACTG2; dysmotility; megacystis-microcolon intestinal hypoperistalsis; smooth muscle; visceral myopathy.
© 2019 Wiley Periodicals, Inc.
Conflict of interest statement
Conflict of Interests
Baylor College of Medicine and Miraca Holdings Inc. have formed a joint venture with shared ownership and governance of Baylor Genetics (BG), formerly the Baylor Miraca Genetics Laboratories, which performs chromosomal microarray analysis and clinical exome sequencing. J.R.L. serves on the Scientific Advisory Board of BG. J.R.L. has stock ownership in 23 and Me, is a paid consultant for Regeneron Pharmaceuticals, and is a coinventor on multiple US and European patents related to molecular diagnostics for inherited neuropathies, eye diseases, and bacterial genomic fingerprinting.
Figures
References
-
- Gauthier J, Ouled Amar Bencheikh B, Hamdan FF, Harrison SM, Baker LA, Couture F, . . . Soucy JF (2015). A homozygous loss-of-function variant in MYH11 in a case with megacystis-microcolon-intestinal hypoperistalsis syndrome. Eur J Hum Genet, 23(9), 1266–1268. doi:10.1038/ejhg.2014.256 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
