

Single-molecule real-time sequencing facilitates the analysis of transcripts and splice isoforms of anthers in Chinese cabbage (Brassica rapa L. ssp. pekinensis)
- PMID: 31771515
- PMCID: PMC6880451
- DOI: 10.1186/s12870-019-2133-z
Single-molecule real-time sequencing facilitates the analysis of transcripts and splice isoforms of anthers in Chinese cabbage (Brassica rapa L. ssp. pekinensis)
Abstract
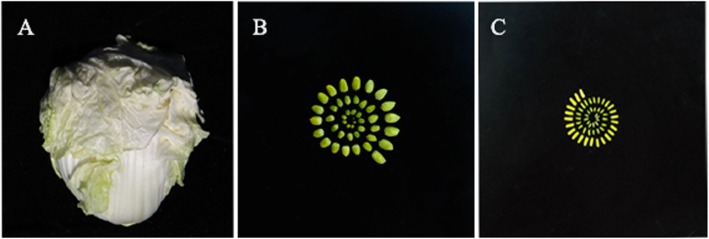
Background: Anther development has been extensively studied at the transcriptional level, but a systematic analysis of full-length transcripts on a genome-wide scale has not yet been published. Here, the Pacific Biosciences (PacBio) Sequel platform and next-generation sequencing (NGS) technology were combined to generate full-length sequences and completed structures of transcripts in anthers of Chinese cabbage.
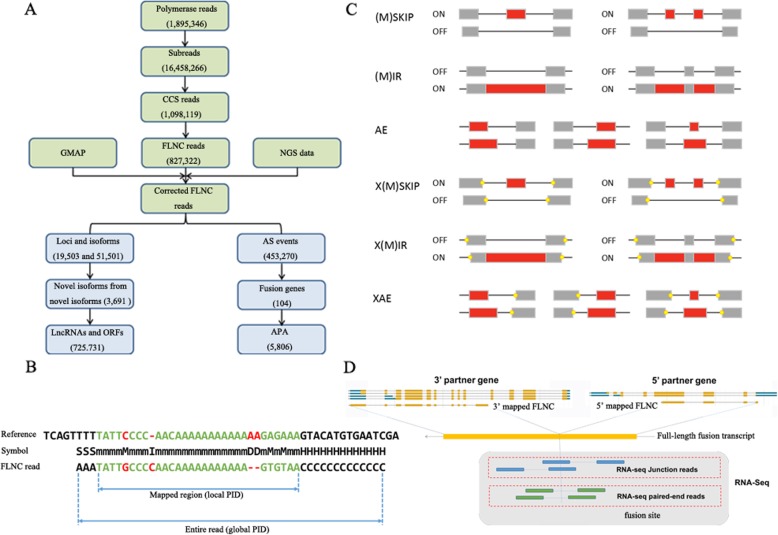
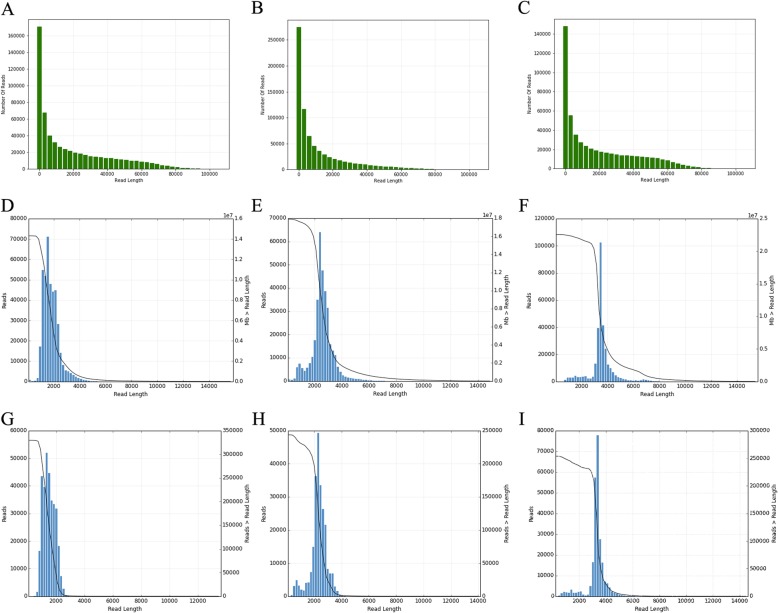
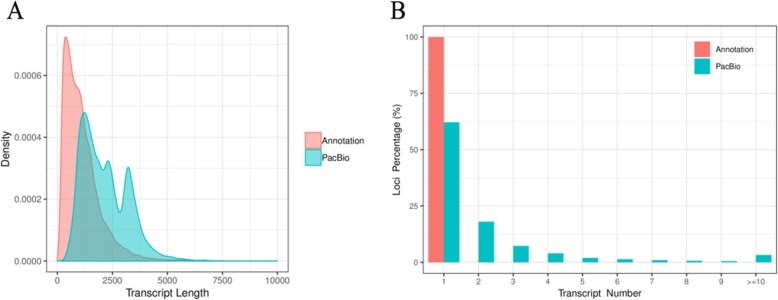
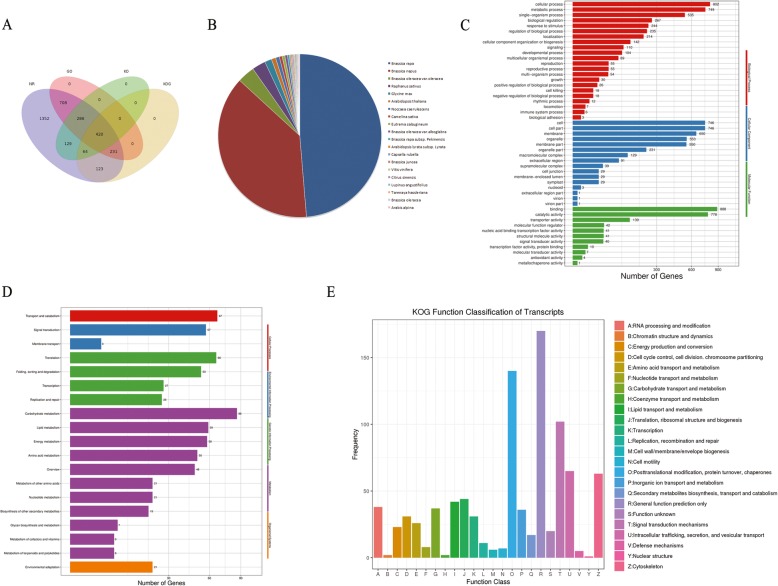
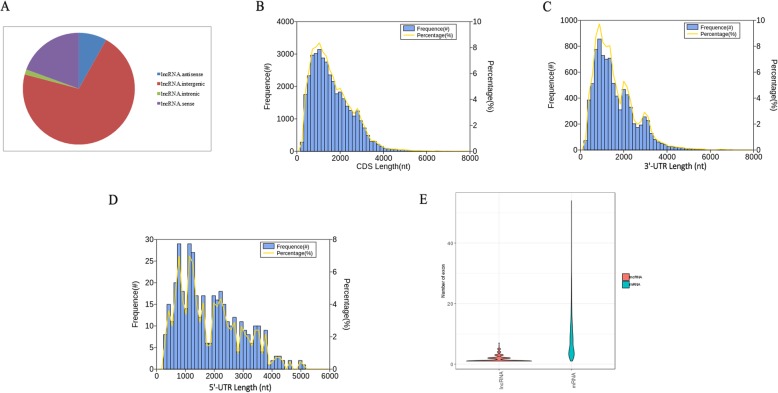
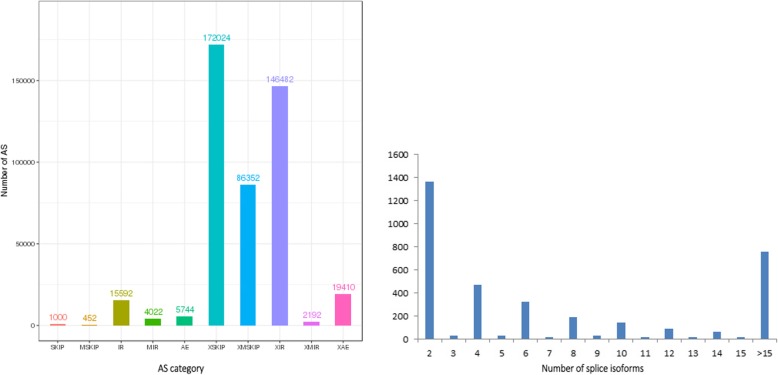
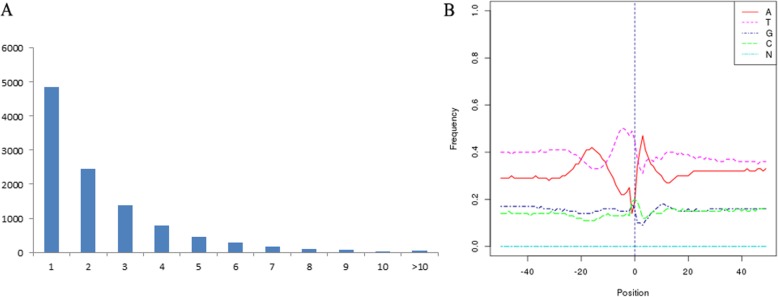
Results: Using single-molecule real-time sequencing (SMRT), a total of 1,098,119 circular consensus sequences (CCSs) were generated with a mean length of 2664 bp. More than 75% of the CCSs were considered full-length non-chimeric (FLNC) reads. After error correction, 725,731 high-quality FLNC reads were estimated to carry 51,501 isoforms from 19,503 loci, consisting of 38,992 novel isoforms from known genes and 3691 novel isoforms from novel genes. Of the novel isoforms, we identified 407 long non-coding RNAs (lncRNAs) and 37,549 open reading frames (ORFs). Furthermore, a total of 453,270 alternative splicing (AS) events were identified and the majority of AS models in anther were determined to be approximate exon skipping (XSKIP) events. Of the key genes regulated during anther development, AS events were mainly identified in the genes SERK1, CALS5, NEF1, and CESA1/3. Additionally, we identified 104 fusion transcripts and 5806 genes that had alternative polyadenylation (APA).
Conclusions: Our work demonstrated the transcriptome diversity and complexity of anther development in Chinese cabbage. The findings provide a basis for further genome annotation and transcriptome research in Chinese cabbage.
Keywords: Alternative splicing; Anther; Chinese cabbage; Full-length transcript; Fusion transcript.
Conflict of interest statement
All the authors declare that they have no competing interests.
Figures


References
-
- Liu C, Liu Z, Li C, Zhang Y, Feng H. Comparative transcriptome analysis of fertile and sterile buds from a genetically male sterile line of Chinese cabbage. In Vitro Cell Dev Biol Plant. 2016;52(2):130–139. doi: 10.1007/s11627-016-9754-9. - DOI
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
