

Gain-of-Function RHOA Mutations Promote Focal Adhesion Kinase Activation and Dependency in Diffuse Gastric Cancer
- PMID: 31771969
- PMCID: PMC7007383
- DOI: 10.1158/2159-8290.CD-19-0811
Gain-of-Function RHOA Mutations Promote Focal Adhesion Kinase Activation and Dependency in Diffuse Gastric Cancer
Abstract
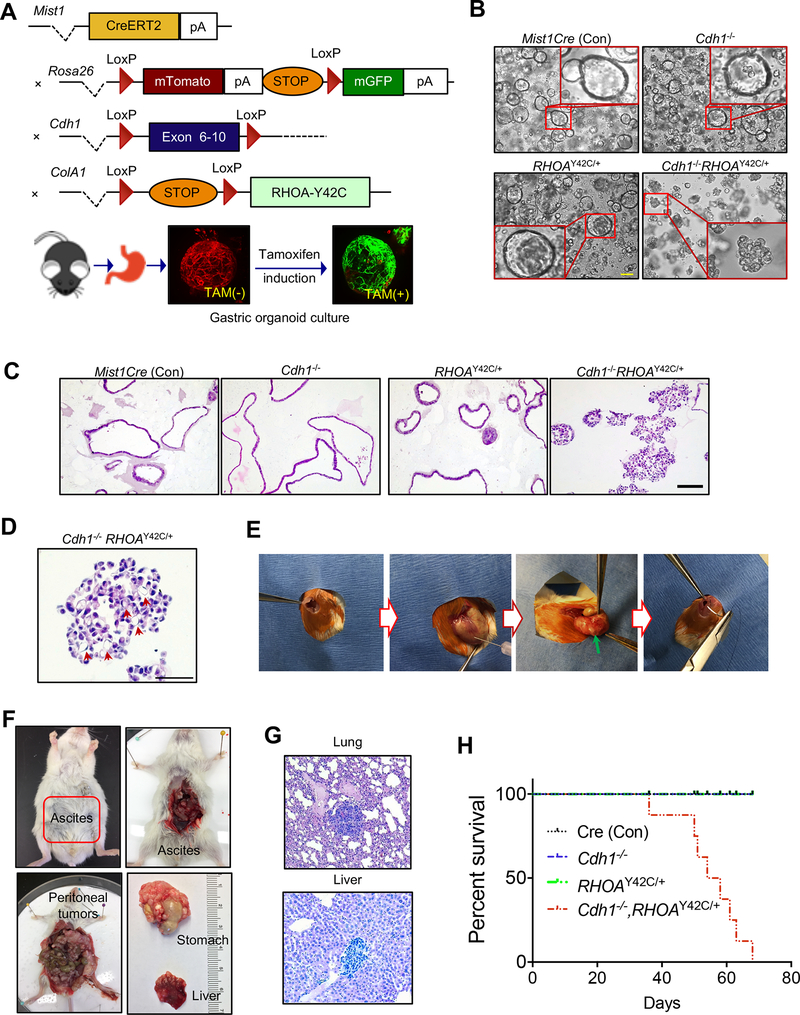
Diffuse gastric cancer (DGC) is a lethal malignancy lacking effective systemic therapy. Among the most provocative recent results in DGC has been that of highly recurrent missense mutations in the GTPase RHOA. The function of these mutations has remained unresolved. We demonstrate that RHOAY42C, the most common RHOA mutation in DGC, is a gain-of-function oncogenic mutant, and that expression of RHOAY42C with inactivation of the canonical tumor suppressor Cdh1 induces metastatic DGC in a mouse model. Biochemically, RHOAY42C exhibits impaired GTP hydrolysis and enhances interaction with its effector ROCK. RHOA Y42C mutation and Cdh1 loss induce actin/cytoskeletal rearrangements and activity of focal adhesion kinase (FAK), which activates YAP-TAZ, PI3K-AKT, and β-catenin. RHOAY42C murine models were sensitive to FAK inhibition and to combined YAP and PI3K pathway blockade. These results, coupled with sensitivity to FAK inhibition in patient-derived DGC cell lines, nominate FAK as a novel target for these cancers. SIGNIFICANCE: The functional significance of recurrent RHOA mutations in DGC has remained unresolved. Through biochemical studies and mouse modeling of the hotspot RHOAY42C mutation, we establish that these mutations are activating, detail their effects upon cell signaling, and define how RHOA-mediated FAK activation imparts sensitivity to pharmacologic FAK inhibitors.See related commentary by Benton and Chernoff, p. 182.This article is highlighted in the In This Issue feature, p. 161.
©2019 American Association for Cancer Research.
Figures






Comment in
-
A New Rho(d) Map to Diffuse Gastric Cancer.Cancer Discov. 2020 Feb;10(2):182-184. doi: 10.1158/2159-8290.CD-19-1327. Cancer Discov. 2020. PMID: 32024682 Free PMC article.
References
-
- Lauren P The Two Histological Main Types of Gastric Carcinoma: Diffuse and So-Called Intestinal-Type Carcinoma. An Attempt at a Histo-Clinical Classification. Acta Pathol Microbiol Scand 1965;64:31–49. - PubMed
-
- Becker KF, Atkinson MJ, Reich U, Becker I, Nekarda H, Siewert JR, et al. E-cadherin gene mutations provide clues to diffuse type gastric carcinomas. Cancer Res 1994;54(14):3845–52. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
