

Evolution and Global Transmission of a Multidrug-Resistant, Community-Associated Methicillin-Resistant Staphylococcus aureus Lineage from the Indian Subcontinent
- PMID: 31772058
- PMCID: PMC6879714
- DOI: 10.1128/mBio.01105-19
Evolution and Global Transmission of a Multidrug-Resistant, Community-Associated Methicillin-Resistant Staphylococcus aureus Lineage from the Indian Subcontinent
Abstract
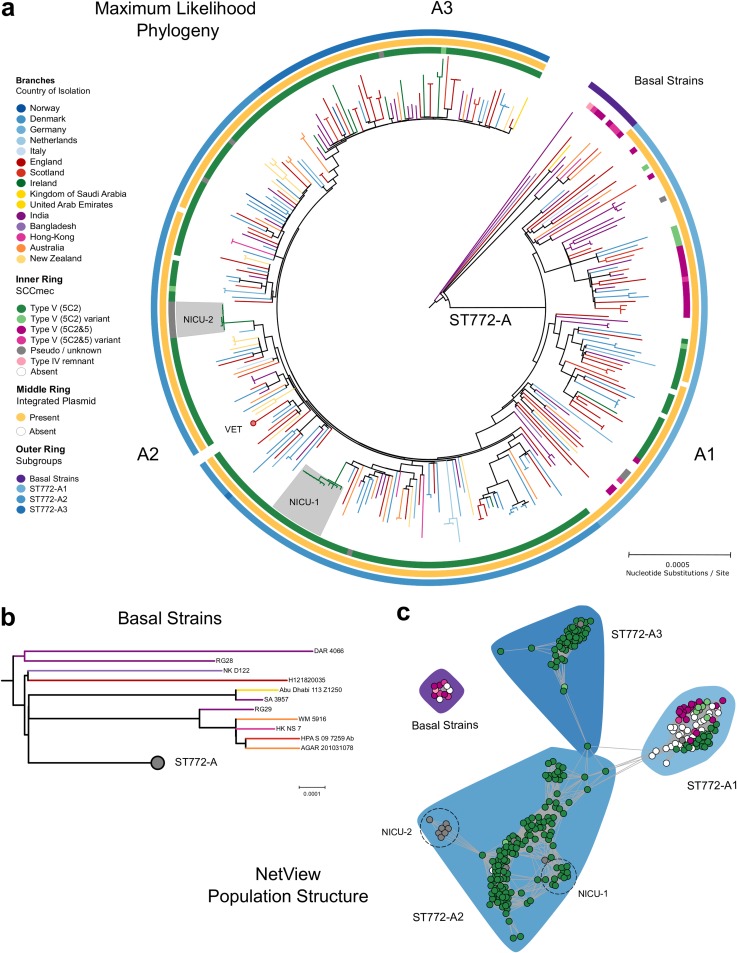
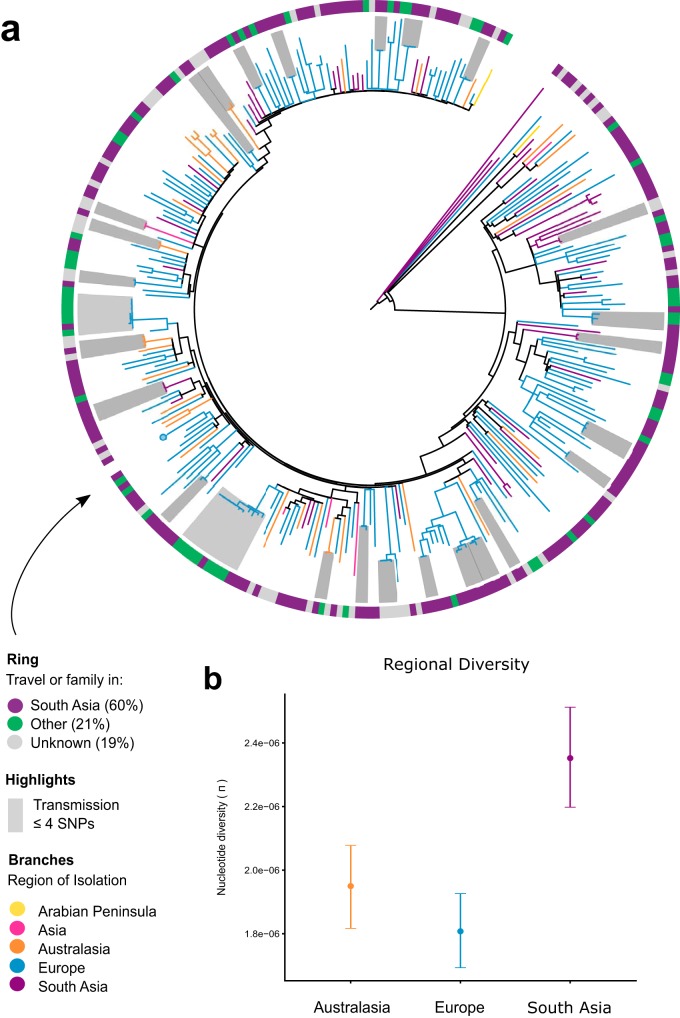
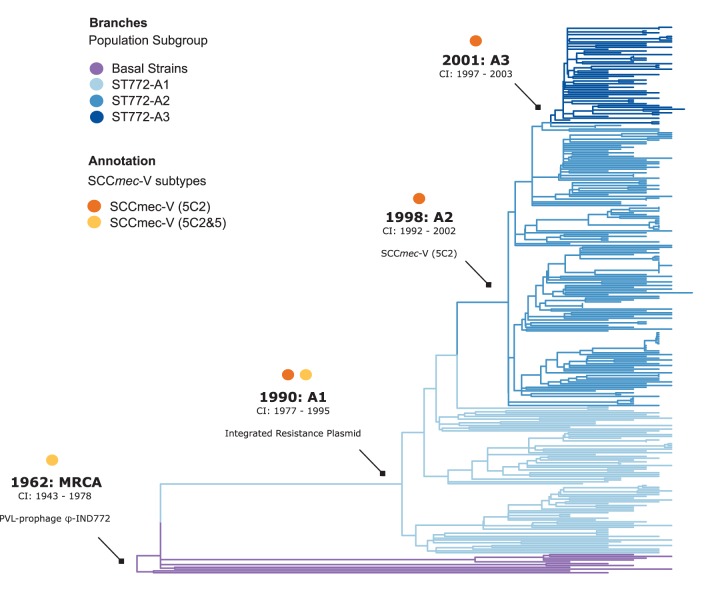
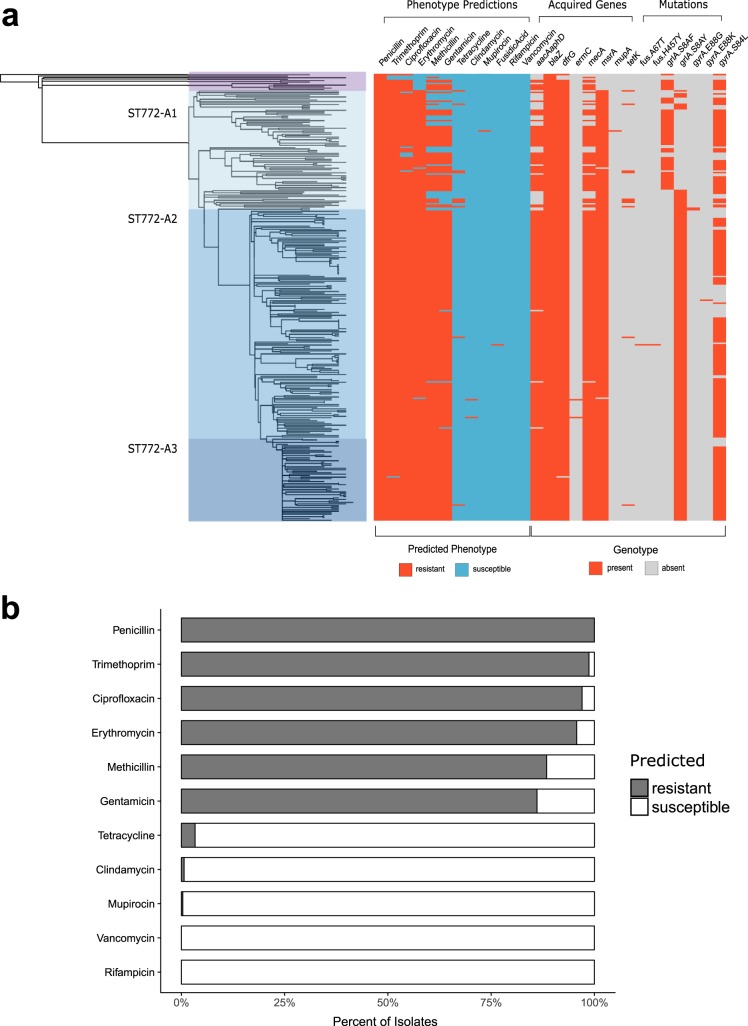
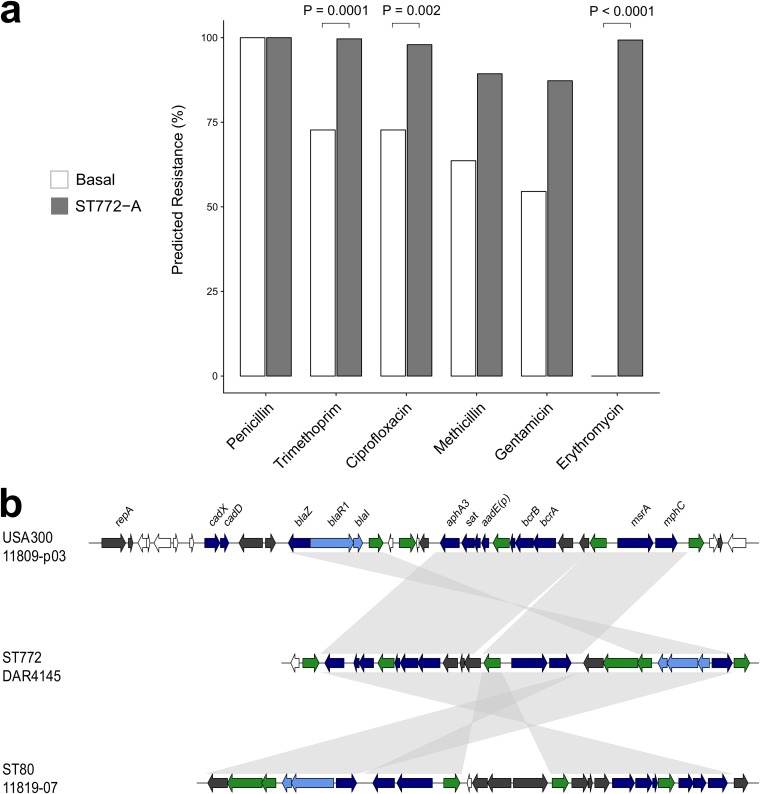
The evolution and global transmission of antimicrobial resistance have been well documented for Gram-negative bacteria and health care-associated epidemic pathogens, often emerging from regions with heavy antimicrobial use. However, the degree to which similar processes occur with Gram-positive bacteria in the community setting is less well understood. In this study, we traced the recent origins and global spread of a multidrug-resistant, community-associated Staphylococcus aureus lineage from the Indian subcontinent, the Bengal Bay clone (ST772). We generated whole-genome sequence data of 340 isolates from 14 countries, including the first isolates from Bangladesh and India, to reconstruct the evolutionary history and genomic epidemiology of the lineage. Our data show that the clone emerged on the Indian subcontinent in the early 1960s and disseminated rapidly in the 1990s. Short-term outbreaks in community and health care settings occurred following intercontinental transmission, typically associated with travel and family contacts on the subcontinent, but ongoing endemic transmission was uncommon. Acquisition of a multidrug resistance integrated plasmid was instrumental in the emergence of a single dominant and globally disseminated clade in the early 1990s. Phenotypic data on biofilm, growth, and toxicity point to antimicrobial resistance as the driving force in the evolution of ST772. The Bengal Bay clone therefore combines the multidrug resistance of traditional health care-associated clones with the epidemiological transmission of community-associated methicillin-resistant S. aureus (MRSA). Our study demonstrates the importance of whole-genome sequencing for tracking the evolution of emerging and resistant pathogens. It provides a critical framework for ongoing surveillance of the clone on the Indian subcontinent and elsewhere.IMPORTANCE The Bengal Bay clone (ST772) is a community-associated and multidrug-resistant Staphylococcus aureus lineage first isolated from Bangladesh and India in 2004. In this study, we showed that the Bengal Bay clone emerged from a virulent progenitor circulating on the Indian subcontinent. Its subsequent global transmission was associated with travel or family contact in the region. ST772 progressively acquired specific resistance elements at limited cost to its fitness and continues to be exported globally, resulting in small-scale community and health care outbreaks. The Bengal Bay clone therefore combines the virulence potential and epidemiology of community-associated clones with the multidrug resistance of health care-associated S. aureus lineages. This study demonstrates the importance of whole-genome sequencing for the surveillance of highly antibiotic-resistant pathogens, which may emerge in the community setting of regions with poor antibiotic stewardship and rapidly spread into hospitals and communities across the world.
Keywords: Bengal Bay; CA-MRSA; India; ST772; South Asia; Staphylococcus aureus; WGS; antimicrobial resistance; genomic epidemiology; global transmission; phenotyping; phylodynamics.
Copyright © 2019 Steinig et al.
Figures
References
-
- Suaya JA, Mera RM, Cassidy A, O’Hara P, Amrine-Madsen H, Burstin S, Miller LG. 2014. Incidence and cost of hospitalizations associated with Staphylococcus aureus skin and soft tissue infections in the United States from 2001 through 2009. BMC Infect Dis 14:296. doi:10.1186/1471-2334-14-296. - DOI - PMC - PubMed
-
- Planet PJ, LaRussa SJ, Dana A, Smith H, Xu A, Ryan C, Uhlemann A-C, Boundy S, Goldberg J, Narechania A, Kulkarni R, Ratner AJ, Geoghegan JA, Kolokotronis S-O, Prince A. 2013. Emergence of the epidemic methicillin-resistant Staphylococcus aureus strain USA300 coincides with horizontal transfer of the arginine catabolic mobile element and speG-mediated adaptations for survival on skin. mBio 4:e00889-13. doi:10.1128/mBio.00889-13. - DOI - PMC - PubMed
-
- Planet PJ, Diaz L, Kolokotronis S-O, Narechania A, Reyes J, Xing G, Rincon S, Smith H, Panesso D, Ryan C, Smith DP, Guzman M, Zurita J, Sebra R, Deikus G, Nolan RL, Tenover FC, Weinstock GM, Robinson DA, Arias CA. 2015. Parallel epidemics of community-associated methicillin-resistant Staphylococcus aureus USA300 infection in North and South America. J Infect Dis 212:1874–1882. doi:10.1093/infdis/jiv320. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
