

Novel TNXB Variants in Two Italian Patients with Classical-Like Ehlers-Danlos Syndrome
- PMID: 31775249
- PMCID: PMC6947605
- DOI: 10.3390/genes10120967
Novel TNXB Variants in Two Italian Patients with Classical-Like Ehlers-Danlos Syndrome
Abstract
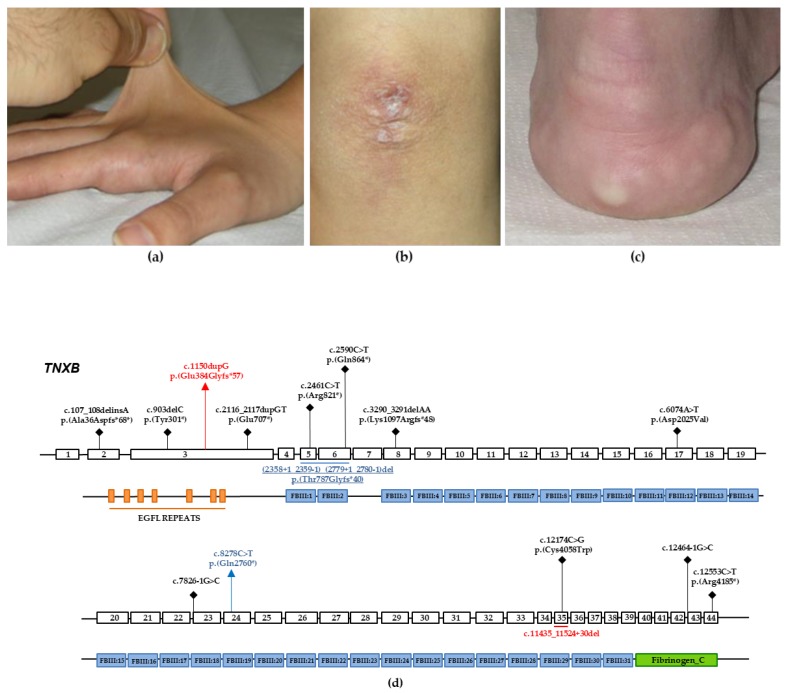
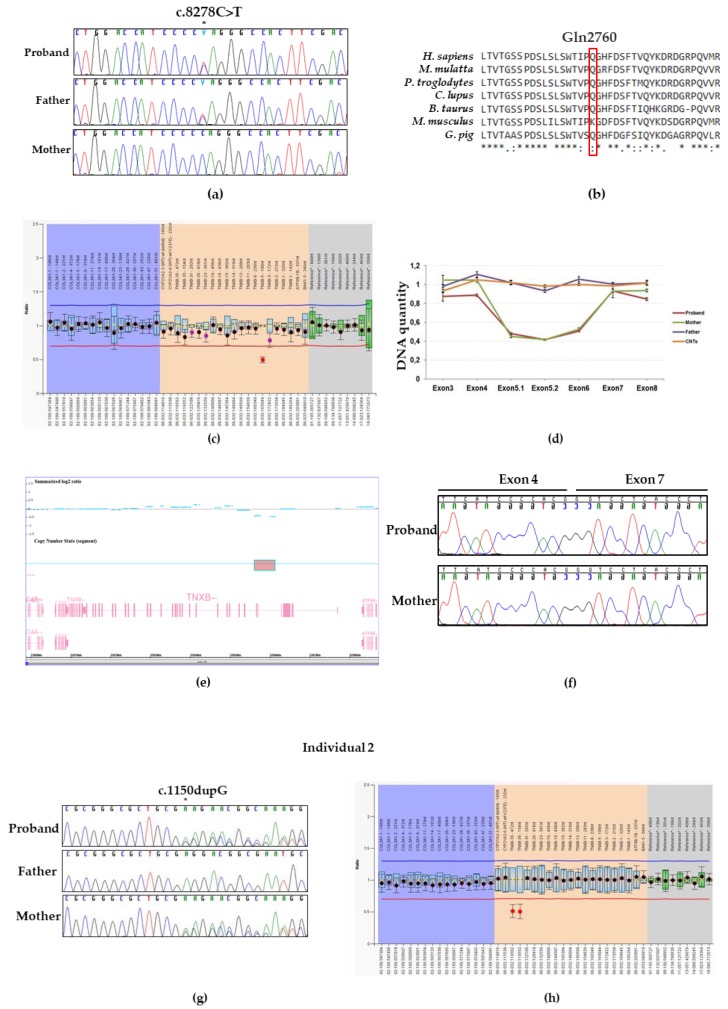
TNXB-related classical-like Ehlers-Danlos syndrome (TNXB-clEDS) is an ultrarare type of Ehlers-Danlos syndrome due to biallelic null variants in TNXB, encoding tenascin-X. Less than 30 individuals have been reported to date, mostly of Dutch origin and showing a phenotype resembling classical Ehlers-Danlos syndrome without atrophic scarring. TNXB-clEDS is likely underdiagnosed due to the complex structure of the TNXB locus, a fact that complicates diagnostic molecular testing. Here, we report two unrelated Italian women with TNXB-clEDS due to compound heterozygosity for null alleles in TNXB. Both presented soft and hyperextensible skin, generalized joint hypermobility and related musculoskeletal complications, and chronic constipation. In addition, individual 1 showed progressive finger contractures and shortened metatarsals, while individual 2 manifested recurrent subconjunctival hemorrhages and an event of spontaneous rupture of the brachial vein. Molecular testing found the two previously unreported c.8278C > T p.(Gln2760*) and the c.(2358 + 1_2359 - 1)_(2779 + 1_2780 - 1)del variants in Individual 1, and the novel c.1150dupG p.(Glu384Glyfs*57) and the recurrent c.11435_11524+30del variants in Individual 2. mRNA analysis confirmed that the c.(2358 + 1_2359 - 1)_(2779 + 1_2780 - 1)del variant causes a frameshift leading to a predicted truncated protein [p.(Thr787Glyfs*40)]. This study refines the phenotype recently delineated in association with biallelic null alleles in TNXB, and adds three novel variants to its mutational repertoire. Unusual digital anomalies seem confirmed as possibly peculiar of TNXB-clEDS, while vascular fragility could be more than a chance association also in this Ehlers-Danlos syndrome type.
Keywords: Classical-like; Ehlers-Danlos syndrome; TNXB; haploinsufficiency; tenascin-X.
Conflict of interest statement
The authors declare no conflict of interest. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Figures
Similar articles
-
TNXB-Related Classical-Like Ehlers-Danlos Syndrome.2022 Sep 15. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2025. 2022 Sep 15. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2025. PMID: 36108117 Free Books & Documents. Review.
-
Clinical and Molecular Characterization of Classical-Like Ehlers-Danlos Syndrome Due to a Novel TNXB Variant.Genes (Basel). 2019 Oct 25;10(11):843. doi: 10.3390/genes10110843. Genes (Basel). 2019. PMID: 31731524 Free PMC article.
-
Recurrent pneumothorax in a case of tenascin-X deficient Ehlers-Danlos syndrome: Broadening the phenotypic spectrum.Am J Med Genet A. 2022 May;188(5):1583-1588. doi: 10.1002/ajmg.a.62674. Epub 2022 Feb 6. Am J Med Genet A. 2022. PMID: 35128805 Free PMC article.
-
An overlapping phenotype of Osteogenesis imperfecta and Ehlers-Danlos syndrome due to a heterozygous mutation in COL1A1 and biallelic missense variants in TNXB identified by whole exome sequencing.Am J Med Genet A. 2016 Apr;170A(4):1080-5. doi: 10.1002/ajmg.a.37547. Epub 2016 Jan 22. Am J Med Genet A. 2016. PMID: 26799614
-
Tenascin-X as a causal gene for classical-like Ehlers-Danlos syndrome.Front Genet. 2023 Mar 15;14:1107787. doi: 10.3389/fgene.2023.1107787. eCollection 2023. Front Genet. 2023. PMID: 37007968 Free PMC article. Review.
Cited by
-
Multisystemic manifestations in a cohort of 75 classical Ehlers-Danlos syndrome patients: natural history and nosological perspectives.Orphanet J Rare Dis. 2020 Jul 31;15(1):197. doi: 10.1186/s13023-020-01470-0. Orphanet J Rare Dis. 2020. PMID: 32736638 Free PMC article.
-
Biomarkers for Ehlers-Danlos Syndromes: There Is a Role?Int J Mol Sci. 2021 Sep 20;22(18):10149. doi: 10.3390/ijms221810149. Int J Mol Sci. 2021. PMID: 34576312 Free PMC article. Review.
-
Genetic diversity assessment and genome-wide association study reveal candidate genes associated with component traits in sweet potato (Ipomoea batatas (L.) Lam).Mol Genet Genomics. 2023 May;298(3):653-667. doi: 10.1007/s00438-023-02007-3. Epub 2023 Mar 21. Mol Genet Genomics. 2023. PMID: 36943475
-
Hypermobile Ehlers-Danlos syndromes: Complex phenotypes, challenging diagnoses, and poorly understood causes.Dev Dyn. 2021 Mar;250(3):318-344. doi: 10.1002/dvdy.220. Epub 2020 Aug 17. Dev Dyn. 2021. PMID: 32629534 Free PMC article. Review.
-
Clinical and molecular delineation of classical-like Ehlers-Danlos syndrome through a comprehensive next-generation sequencing-based screening system.Front Genet. 2023 Aug 30;14:1234804. doi: 10.3389/fgene.2023.1234804. eCollection 2023. Front Genet. 2023. PMID: 37712068 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
