

Unraveling cis and trans regulatory evolution during cotton domestication
- PMID: 31776348
- PMCID: PMC6881400
- DOI: 10.1038/s41467-019-13386-w
Unraveling cis and trans regulatory evolution during cotton domestication
Abstract
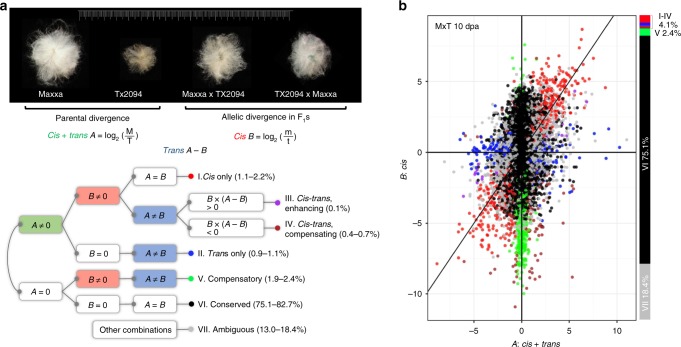
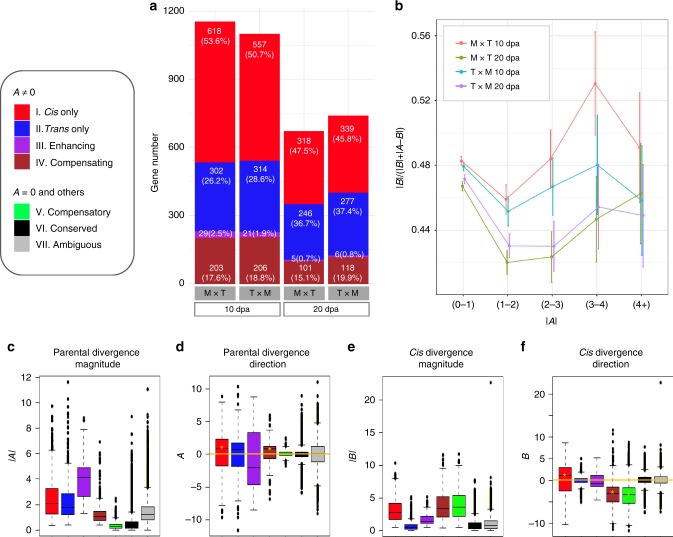
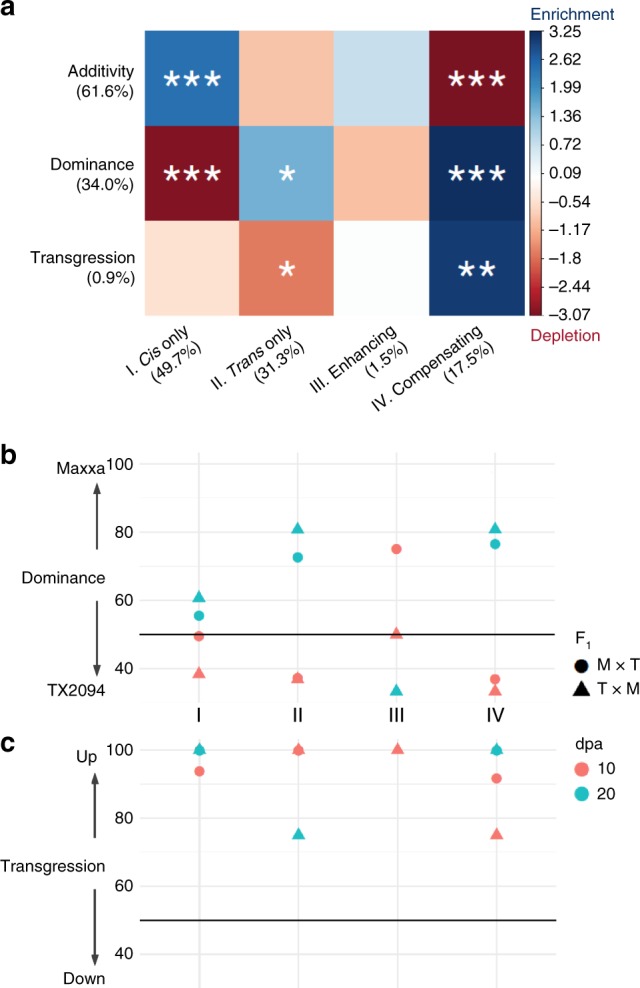
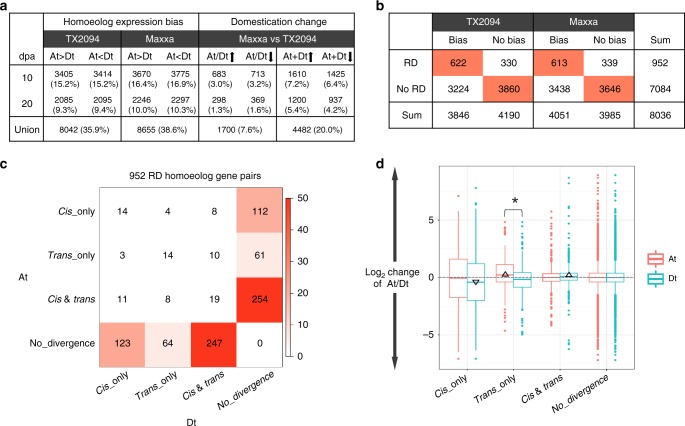
Cis and trans regulatory divergence underlies phenotypic and evolutionary diversification. Relatively little is understood about the complexity of regulatory evolution accompanying crop domestication, particularly for polyploid plants. Here, we compare the fiber transcriptomes between wild and domesticated cotton (Gossypium hirsutum) and their reciprocal F1 hybrids, revealing genome-wide (~15%) and often compensatory cis and trans regulatory changes under divergence and domestication. The high level of trans evolution (54%-64%) observed is likely enabled by genomic redundancy following polyploidy. Our results reveal that regulatory variation is significantly associated with sequence evolution, inheritance of parental expression patterns, co-expression gene network properties, and genomic loci responsible for domestication traits. With respect to regulatory evolution, the two subgenomes of allotetraploid cotton are often uncoupled. Overall, our work underscores the complexity of regulatory evolution during fiber domestication and may facilitate new approaches for improving cotton and other polyploid plants.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
