

Chromatin structure dynamics during the mitosis-to-G1 phase transition
- PMID: 31776509
- PMCID: PMC6895436
- DOI: 10.1038/s41586-019-1778-y
Chromatin structure dynamics during the mitosis-to-G1 phase transition
Abstract
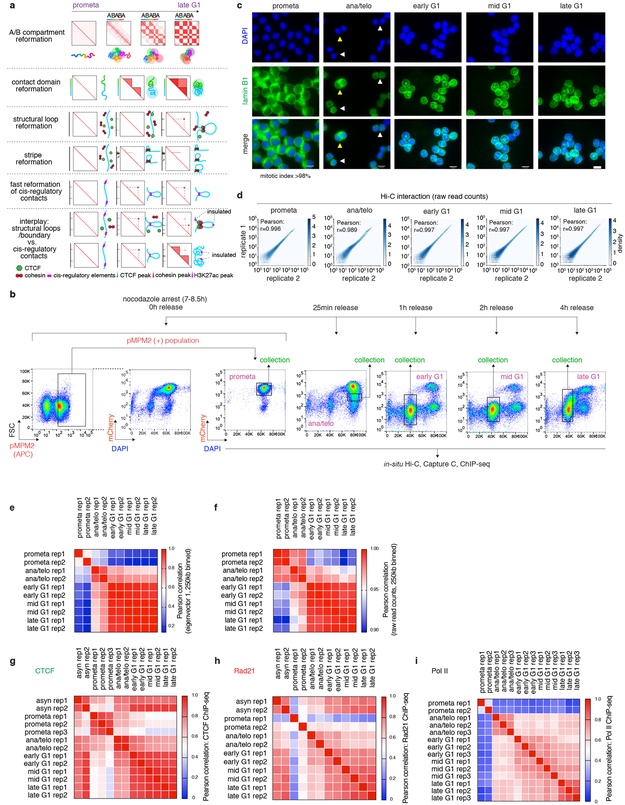
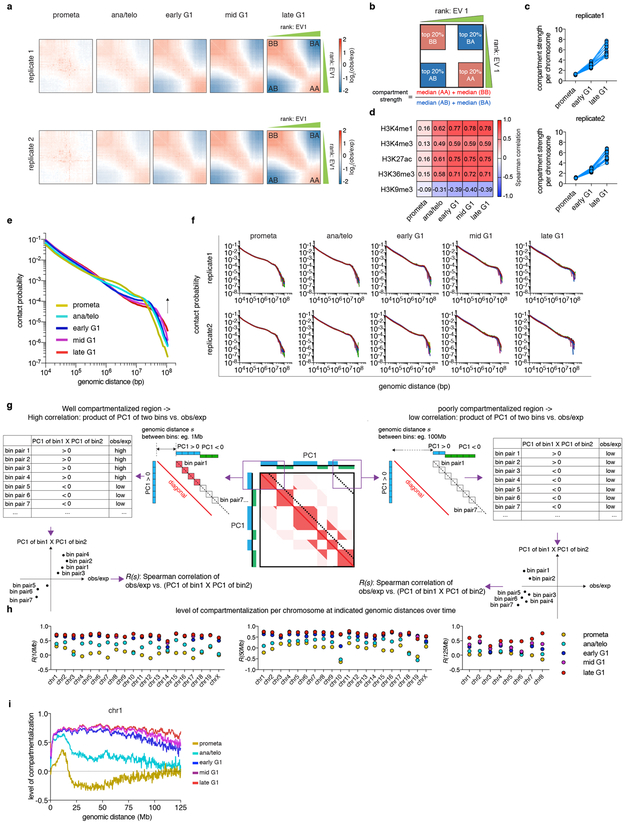
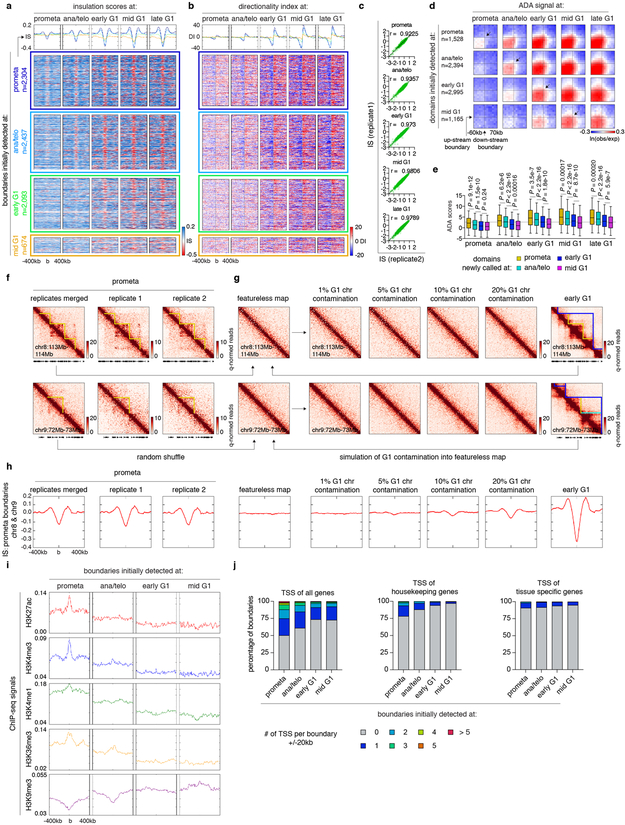
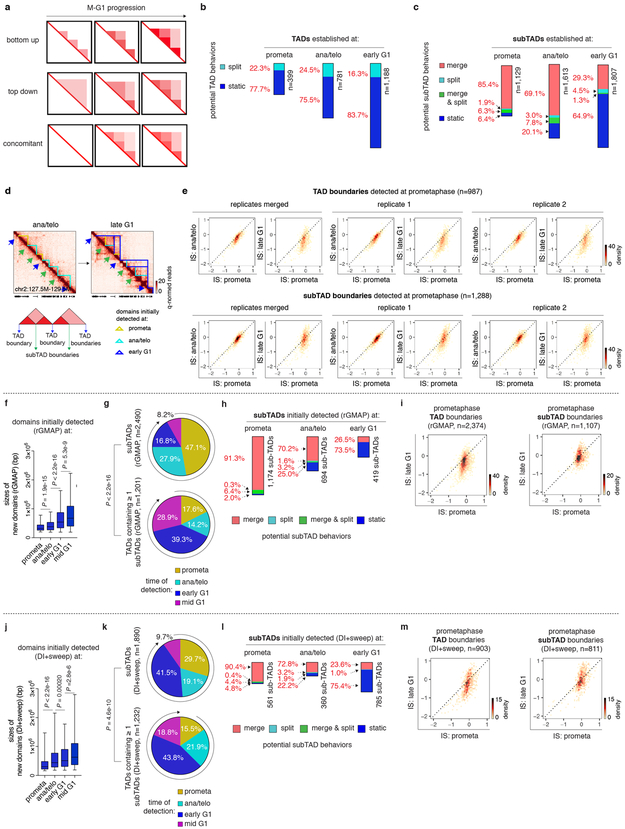
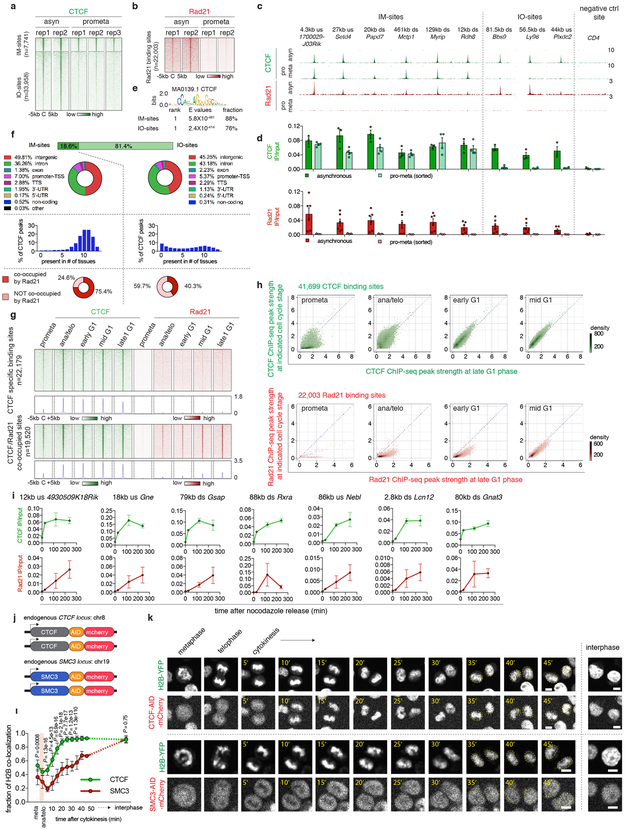
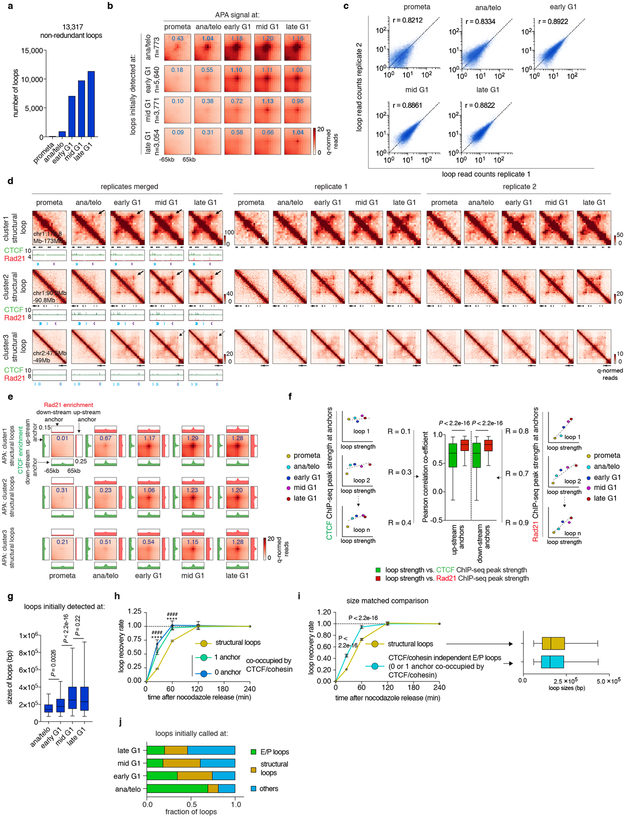
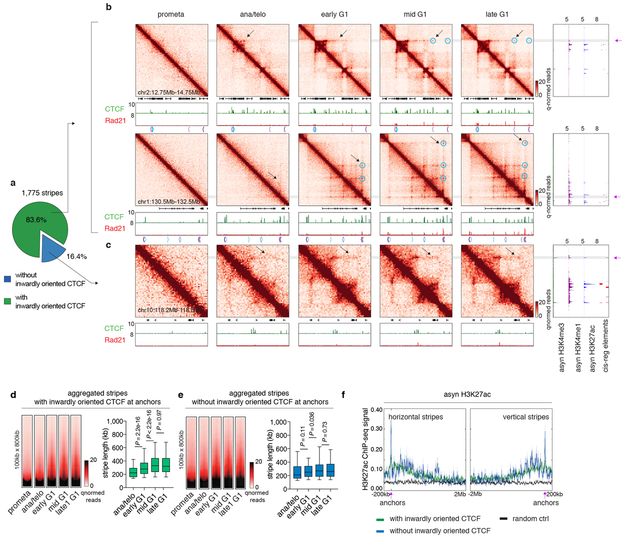
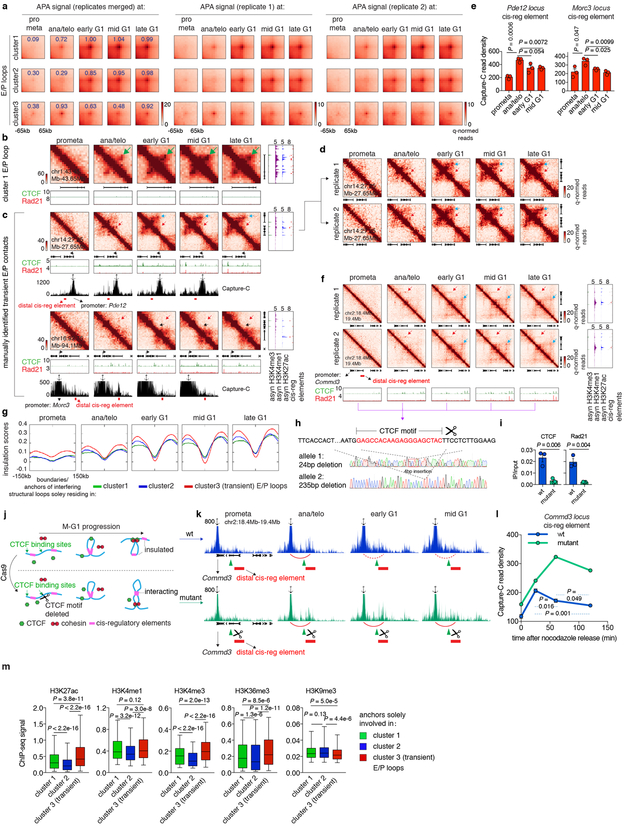
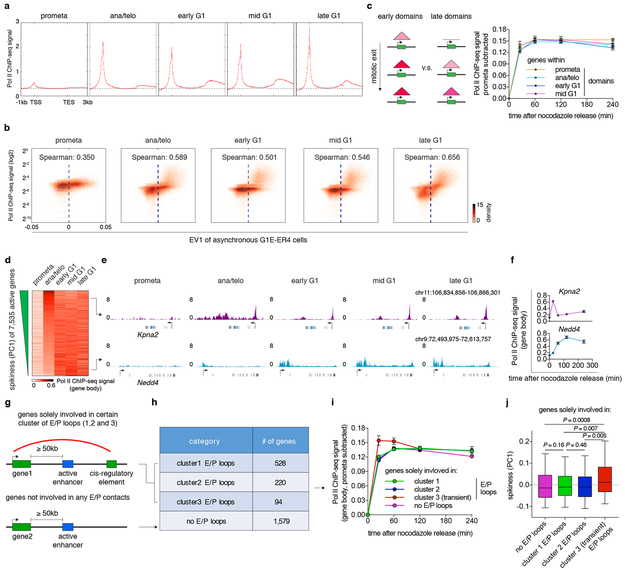
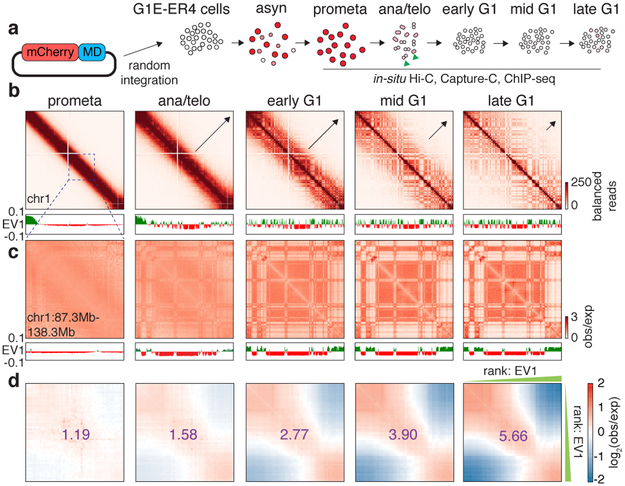
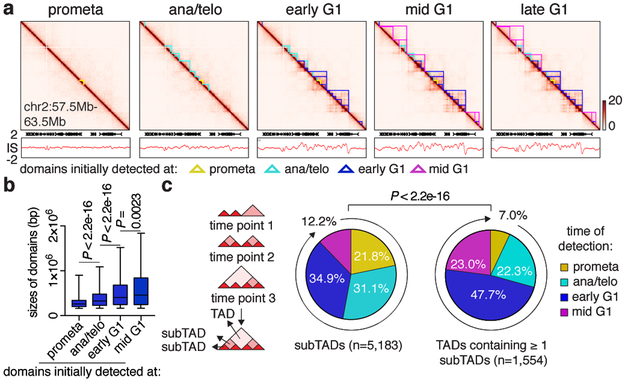
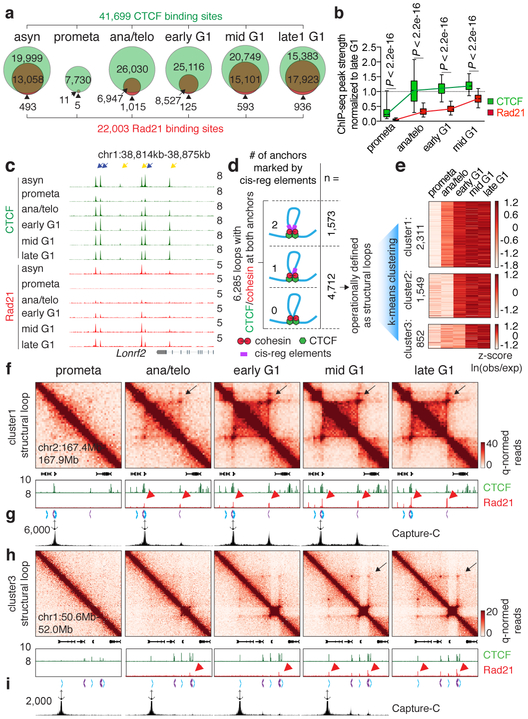
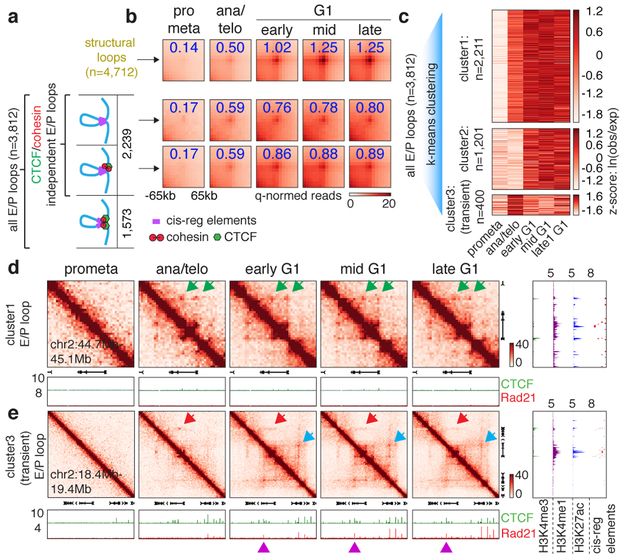
Features of higher-order chromatin organization-such as A/B compartments, topologically associating domains and chromatin loops-are temporarily disrupted during mitosis1,2. Because these structures are thought to influence gene regulation, it is important to understand how they are re-established after mitosis. Here we examine the dynamics of chromosome reorganization by Hi-C after mitosis in highly purified, synchronous mouse erythroid cell populations. We observed rapid establishment of A/B compartments, followed by their gradual intensification and expansion. Contact domains form from the 'bottom up'-smaller subTADs are formed initially, followed by convergence into multi-domain TAD structures. CTCF is partially retained on mitotic chromosomes and immediately resumes full binding in ana/telophase. By contrast, cohesin is completely evicted from mitotic chromosomes and regains focal binding at a slower rate. The formation of CTCF/cohesin co-anchored structural loops follows the kinetics of cohesin positioning. Stripe-shaped contact patterns-anchored by CTCF-grow in length, which is consistent with a loop-extrusion process after mitosis. Interactions between cis-regulatory elements can form rapidly, with rates exceeding those of CTCF/cohesin-anchored contacts. Notably, we identified a group of rapidly emerging transient contacts between cis-regulatory elements in ana/telophase that are dissolved upon G1 entry, co-incident with the establishment of inner boundaries or nearby interfering chromatin loops. We also describe the relationship between transcription reactivation and architectural features. Our findings indicate that distinct but mutually influential forces drive post-mitotic chromatin reconfiguration.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
