

The NLRP3 p.A441V Mutation in NLRP3-AID Pathogenesis: Functional Consequences, Phenotype-Genotype Correlations and Evidence for a Recurrent Mutational Event
- PMID: 31777803
- PMCID: PMC6857991
- DOI: 10.1002/acr2.1039
The NLRP3 p.A441V Mutation in NLRP3-AID Pathogenesis: Functional Consequences, Phenotype-Genotype Correlations and Evidence for a Recurrent Mutational Event
Abstract
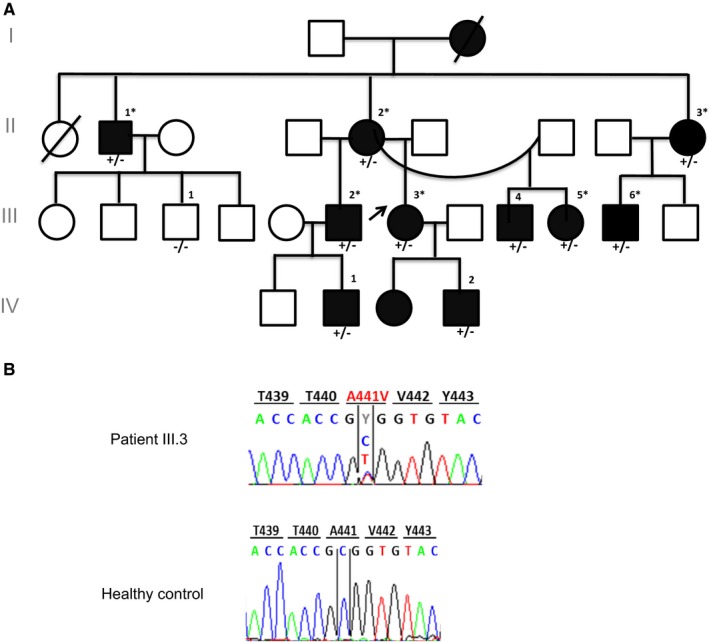
Objective: To determine the molecular and cellular bases of autoinflammatory syndromes in a multigenerational French family with Muckle-Wells syndrome and in a patient originating from Portugal with familial cold autoinflammatory syndrome.
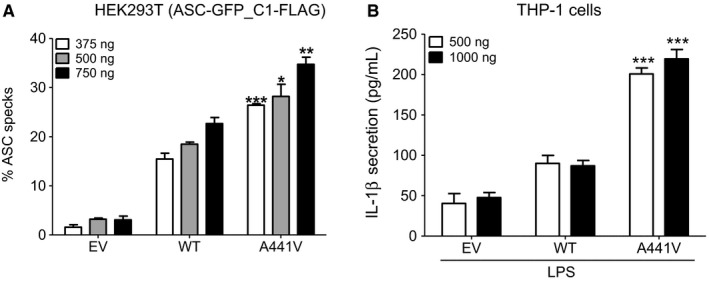
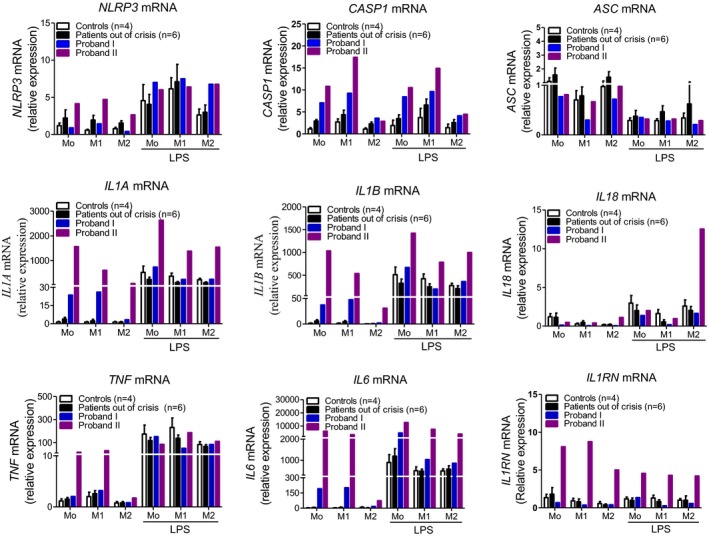
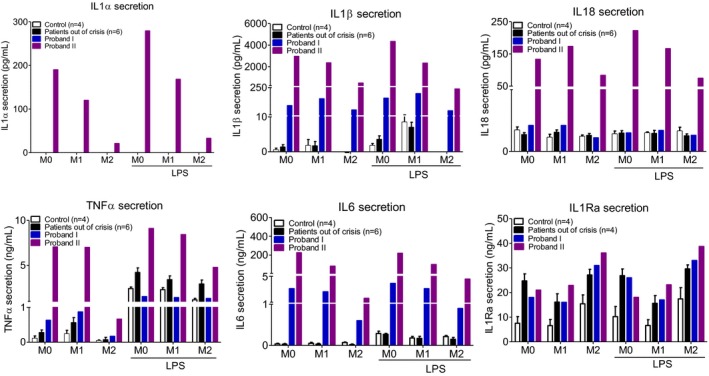
Methods: Sequencing of NLRP3 exon 3 was performed in all accessible patients. Microsatellite and whole-genome single nucleotide polymorphism genotyping was used i) to test the intrafamilial segregation of the identified variant and ii) to look for a founder effect. Functional analyses included the study of i) apoptosis-associated speck-like protein containing a CARD (ASC) speck formation in HEK293T cells (stably expressing ASC-green fluorescent protein and pro-caspase 1-FLAG) transiently expressing the wild-type or mutated NLRP3 protein, ii) levels of IL-1β secreted from transfected THP-1 cells, and iii) inflammasome-related gene expression and cytokine secretion from monocytes isolated from patients in crisis (probands from the two families), related patients out of crisis, and from controls.
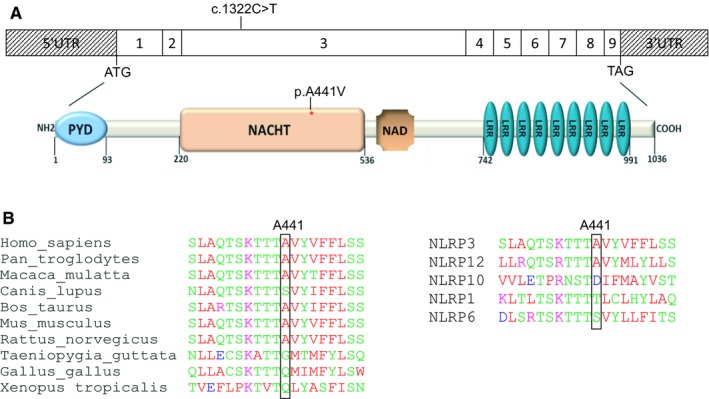
Results: The same heterozygous mutation (c.1322C>T, p.A441V) located in the NACHT domain, segregating with the disease within the first family, was identified in the two families. This mutation was found to be associated with different core haplotypes. NLRP3-A441V led to increased ASC speck formation and high levels of secreted IL-1β. Monocyte inflammasome-related gene expression and cytokine secretion, which were within the normal range in patients out of crisis, were found to be differentially regulated between the two probands, correlating with their phenotypic status.
Conclusion: These molecular and cellular findings, which indicate a recurrent mutational event, clearly demonstrate the pathogenicity of the p.A441V missense mutation in NLRP3-associated autoinflammatory disease and point to the interest of studying patients' primary cells to assess disease activity.
Keywords: IL‐1β; NLRP3; NLRP3‐AID; cryopyrin‐associated periodic syndromes; inflammasome; variation.
© 2019 The Authors. ACR Open Rheumatology published by Wiley Periodicals, Inc. on behalf of American College of Rheumatology.
Figures
References
-
- Ben‐Chetrit E, Gattorno M, Gul A, Kastner DL, Lachmann HJ, Touitou I, et al. Consensus proposal for taxonomy and definition of the autoinflammatory diseases (AIDs): a Delphi study. Ann Rheum Dis 2018;77:1558–65. - PubMed
-
- Kuemmerle‐Deschner JB. CAPS–pathogenesis, presentation and treatment of an autoinflammatory disease. Semin Immunopathol 2015;37:377–85. - PubMed
-
- Martinon F, Mayor A, Tschopp J. The inflammasomes: guardians of the body. Annu Rev Immunol 2009;27:229–65. - PubMed
LinkOut - more resources
Full Text Sources
Miscellaneous
