

Heterozygous Deletion of the SHOX Gene Enhancer in two Females With Clinical Heterogeneity Associating With Skewed XCI and Escaping XCI
- PMID: 31781162
- PMCID: PMC6852097
- DOI: 10.3389/fgene.2019.01086
Heterozygous Deletion of the SHOX Gene Enhancer in two Females With Clinical Heterogeneity Associating With Skewed XCI and Escaping XCI
Abstract
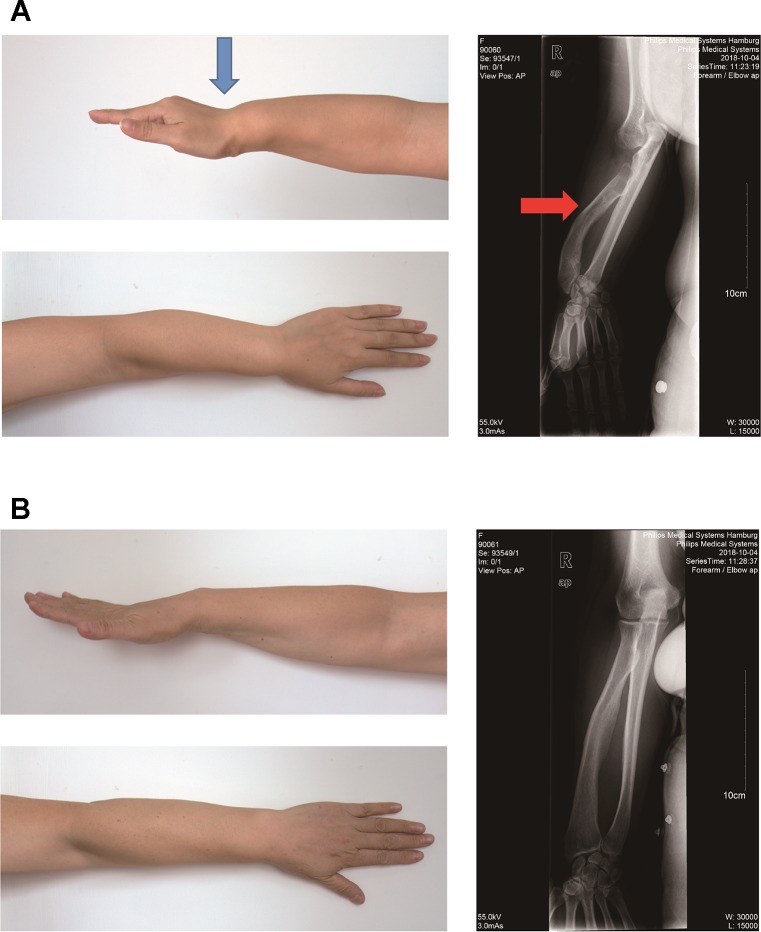
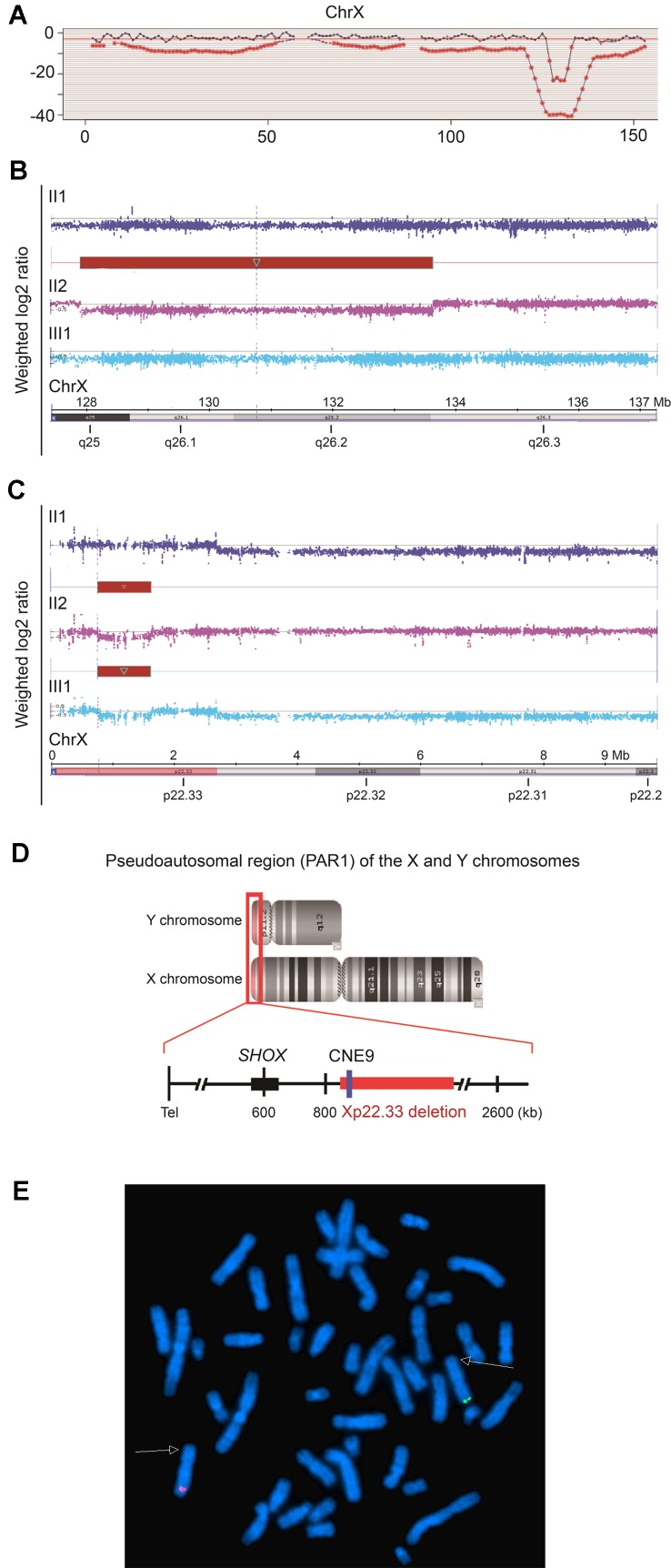
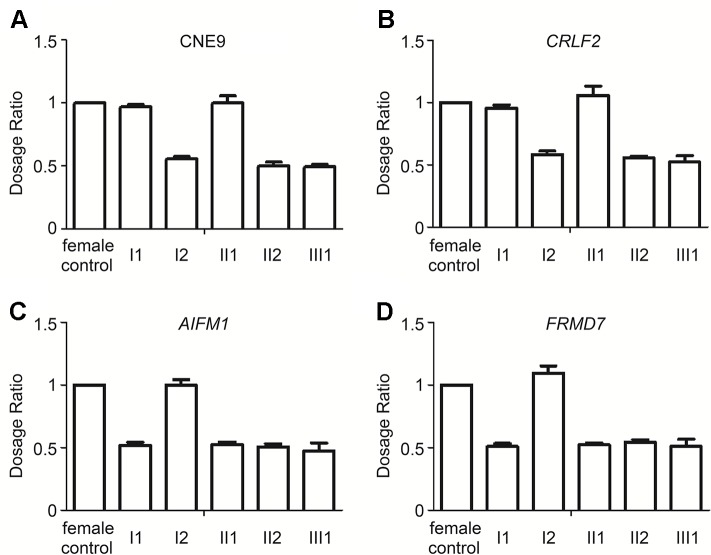
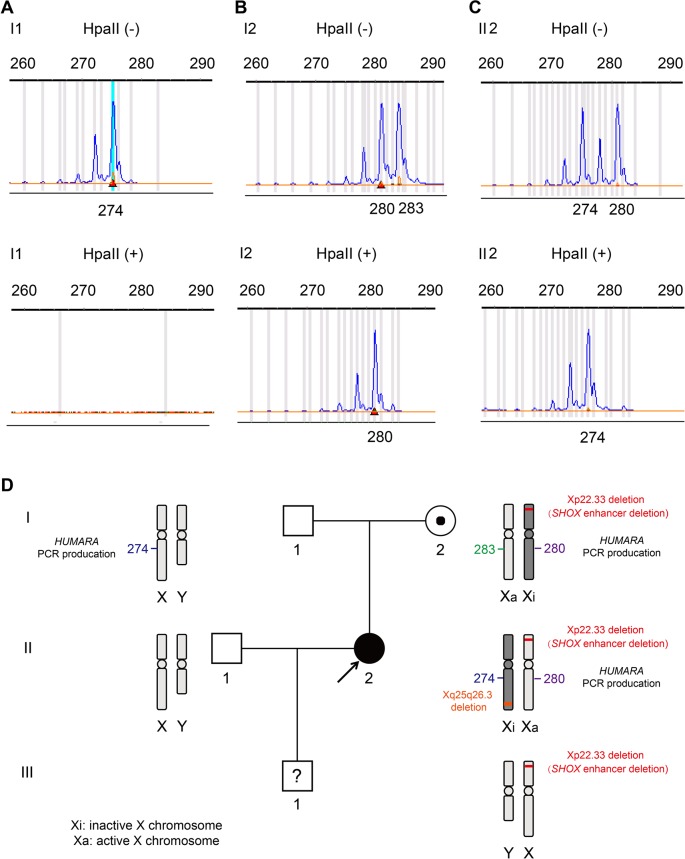
Skewed X-chromosome inactivation (XCI) plays an important role in the phenotypic heterogeneity of X-linked disorders. However, the role of skewed XCI in XCI-escaping gene SHOX regulation is unclear. Here, we focused on a heterozygous deletion of SHOX gene enhancer with clinical heterogeneity. Using SNP array, we detected that the female proband with Leri-Weill dyschondrosteosis (LWD) carried an 857 kb deletion on Xp22.3 (encompassing SHOX enhancer) and a 5,707 kb large-fragment deletion on Xq25q26. XCI analysis revealed that the X-chromosome with the Xq25q26 large-fragment deletion was completely inactivated, which forced the complete activation of the other X-chromosome carrying SHOX enhancer deletion. While the Xp22.3 deletion locates on the escaping XCI region, under the combined action of skewed XCI and escaping XCI, transcription of SHOX gene was mainly from the activated X-chromosome with SHOX enhancer defect, involving in the formation of LWD phenotype. Interestingly, this SHOX enhancer deletion was inherited from her healthy mother, who also demonstrated completely skewed XCI. However, the X-chromosome with SHOX enhancer deletion was inactivated, and the normal X-chromosome was activated. Combing with escaping XCI, her phenotype was almost normal. In summary, this study was a rare report of SHOX gene enhancer deletion in a family with clinical heterogeneity due to skewed inactivation of different X-chromosomes, which can help in the genetic counseling and prenatal diagnosis of disorders in females with SHOX defect.
Keywords: HUMARA assay; Leri-Weill dyschondrosteosis; SHOX gene enhancer; clinical heterogeneity; escaping X-chromosome inactivation (XCI); skewed X-chromosome inactivation (XCI).
Copyright © 2019 Sun, Luo, Qian, Chen, Wang, Li, Zou and Dong.
Figures
Similar articles
-
Case Report: De novo DDX3X mutation caused intellectual disability in a female with skewed X-chromosome inactivation on the mutant allele.Front Genet. 2022 Oct 10;13:999442. doi: 10.3389/fgene.2022.999442. eCollection 2022. Front Genet. 2022. PMID: 36299587 Free PMC article.
-
Identification of the first recurrent PAR1 deletion in Léri-Weill dyschondrosteosis and idiopathic short stature reveals the presence of a novel SHOX enhancer.J Med Genet. 2012 Jul;49(7):442-50. doi: 10.1136/jmedgenet-2011-100678. J Med Genet. 2012. PMID: 22791839
-
Microdeletion in the SHOX 3' region associated with skeletal phenotypes of Langer mesomelic dysplasia in a 45,X/46,X,r(X) infant and Leri-Weill dyschondrosteosis in her 46,XX mother: implication for the SHOX enhancer.Am J Med Genet A. 2005 Aug 15;137(1):72-6. doi: 10.1002/ajmg.a.30852. Am J Med Genet A. 2005. PMID: 16007631 Review.
-
PAR1 deletions downstream of SHOX are the most frequent defect in a Spanish cohort of Léri-Weill dyschondrosteosis (LWD) probands.Hum Mutat. 2006 Oct;27(10):1062. doi: 10.1002/humu.9456. Hum Mutat. 2006. PMID: 16941489
-
[From gene to disease; from SHOX to Lèri-Weill dyschondrosteosis, Turner syndrome and idiopathic short stature].Ned Tijdschr Geneeskd. 2001 Jul 28;145(30):1456-9. Ned Tijdschr Geneeskd. 2001. PMID: 11503314 Review. Dutch.
Cited by
-
Case Report: De novo DDX3X mutation caused intellectual disability in a female with skewed X-chromosome inactivation on the mutant allele.Front Genet. 2022 Oct 10;13:999442. doi: 10.3389/fgene.2022.999442. eCollection 2022. Front Genet. 2022. PMID: 36299587 Free PMC article.
-
Double Isochromosome X, a Rare Cytogenetic Variant of Turner Syndrome: A Case Report and a Review of the Literature.Balkan J Med Genet. 2023 Mar 1;25(1):101-104. doi: 10.2478/bjmg-2022-0011. eCollection 2022 Jun. Balkan J Med Genet. 2023. PMID: 36880033 Free PMC article.
-
Mild phenotypes in patients with different deletions in the 3' enhancer region of SHOX.Eur J Hum Genet. 2024 Jun 24. doi: 10.1038/s41431-024-01646-3. Online ahead of print. Eur J Hum Genet. 2024. PMID: 38914686
-
Beyond the exome: What's next in diagnostic testing for Mendelian conditions.Am J Hum Genet. 2023 Aug 3;110(8):1229-1248. doi: 10.1016/j.ajhg.2023.06.009. Am J Hum Genet. 2023. PMID: 37541186 Free PMC article. Review.
-
Beyond the exome: what's next in diagnostic testing for Mendelian conditions.ArXiv [Preprint]. 2023 Jan 18:arXiv:2301.07363v1. ArXiv. 2023. Update in: Am J Hum Genet. 2023 Aug 3;110(8):1229-1248. doi: 10.1016/j.ajhg.2023.06.009. PMID: 36713248 Free PMC article. Updated. Preprint.
References
-
- Benito-Sanz S., Royo J. L., Barroso E., Paumard-Hernandez B., Barreda-Bonis A. C., Liu P., et al. (2012). Identification of the first recurrent PAR1 deletion in Leri-Weill dyschondrosteosis and idiopathic short stature reveals the presence of a novel SHOX enhancer. J. Med. Genet. 49 (7), 442–450. 10.1136/jmedgenet-2011-100678 - DOI - PubMed
-
- Benito-Sanz S., Thomas N. S., Huber C., Gorbenko del Blanco D., Aza-Carmona M., Crolla J. A., et al. (2005). A novel class of Pseudoautosomal region 1 deletions downstream of SHOX is associated with Leri-Weill dyschondrosteosis. Am. J. Hum. Genet. 77 (4), 533–544. 10.1086/449313 - DOI - PMC - PubMed
-
- Binder G., Rappold G. A. (1993). “SHOX Deficiency Disorders,” in GeneReviews((R)). Eds. Adam M. P., Ardinger H. H., Pagon R. A., Wallace S. E., Bean L. J. H., Stephens K., Amemiya A.(Seattle (WA: : University of Washington; )). - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Molecular Biology Databases