

Spastin MIT Domain Disease-Associated Mutations Disrupt Lysosomal Function
- PMID: 31787869
- PMCID: PMC6856053
- DOI: 10.3389/fnins.2019.01179
Spastin MIT Domain Disease-Associated Mutations Disrupt Lysosomal Function
Abstract
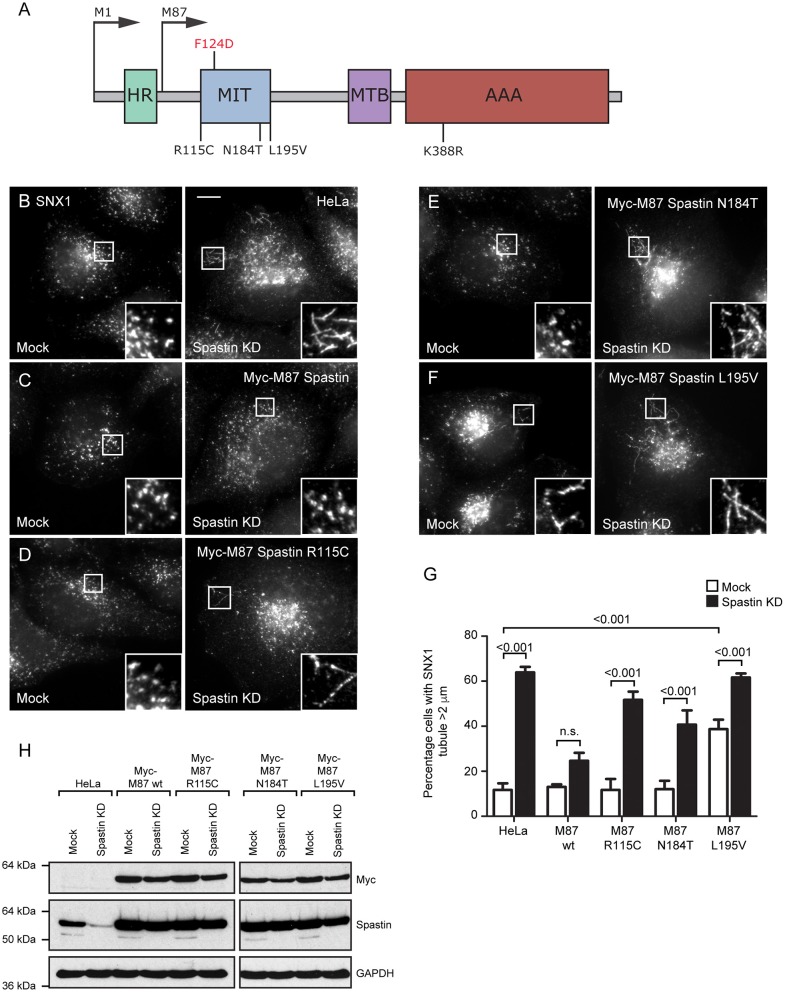
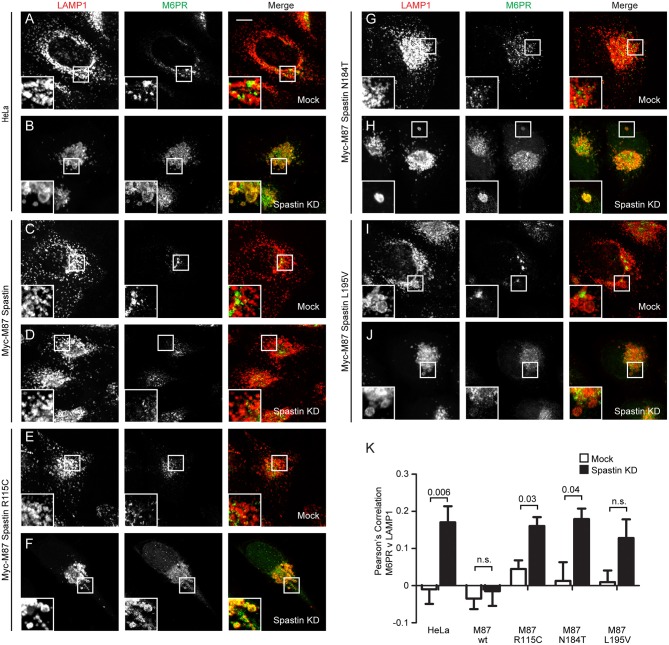
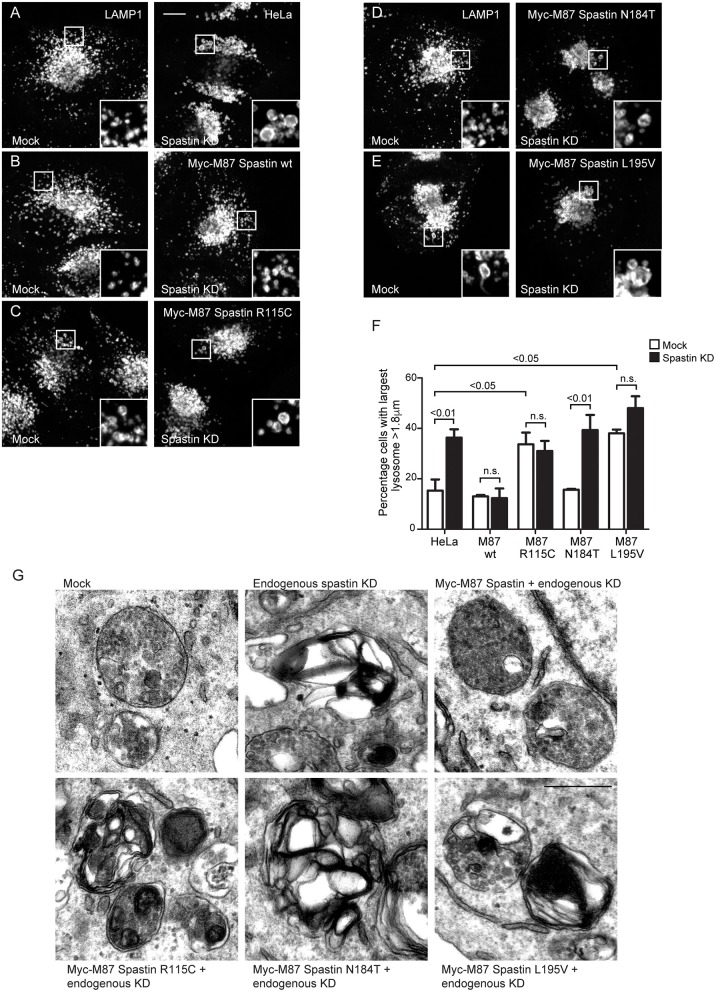
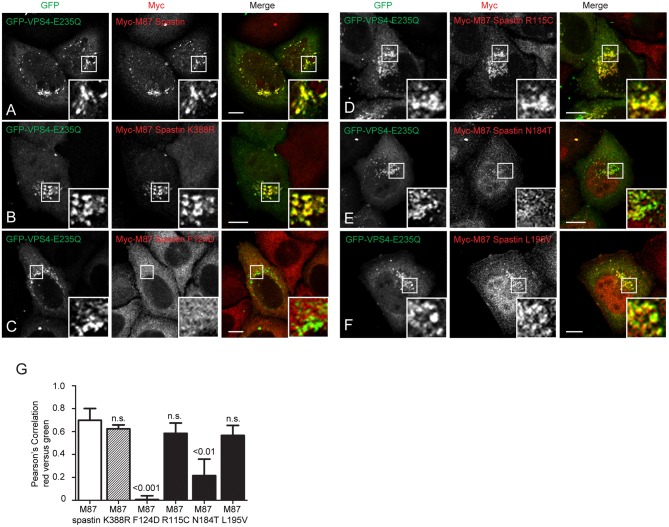
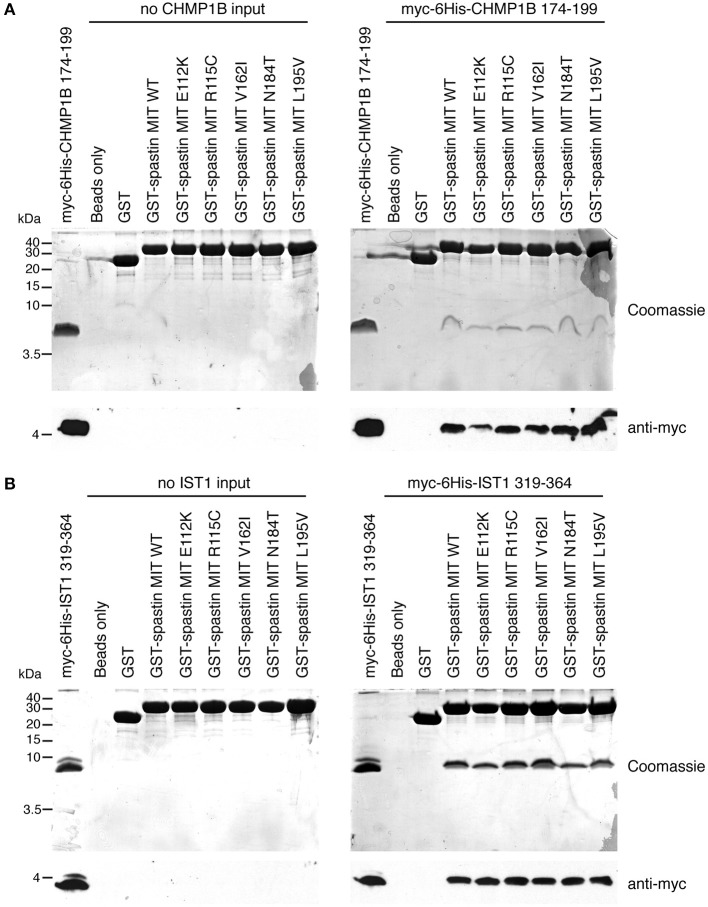
The hereditary spastic paraplegias (HSPs) are genetic motor neuron diseases characterized by progressive degeneration of corticospinal tract axons. Mutations in SPAST, encoding the microtubule-severing ATPase spastin, are the most common causes of HSP. The broad SPAST mutational spectrum indicates a haploinsufficiency pathogenic mechanism in most cases. Most missense mutations cluster in the ATPase domain, where they disrupt the protein's ability to sever microtubules. However, several putative missense mutations in the protein's microtubule interacting and trafficking (MIT) domain have also been described, but the pathogenicity of these mutations has not been verified with functional studies. Spastin promotes endosomal tubule fission, and defects in this lead to lysosomal enzyme mistrafficking and downstream lysosomal abnormalities. We investigated the function of three disease-associated spastin MIT mutants and found that none was able to promote normal endosomal tubule fission, lysosomal enzyme receptor trafficking, or lysosomal morphology. One of the mutations affected recruitment of spastin to endosomes, a property that requires the canonical function of the MIT domain in binding endosomal sorting complex required for transport (ESCRT)-III proteins. However, the other mutants did not affect spastin's endosomal recruitment, raising the possibility of pathologically important non-canonical roles for the MIT domain. In conclusion, we demonstrate that spastin MIT mutants cause functional abnormalities related to the pathogenesis of HSP. These mutations do not directly affect spastin's microtubule-severing capacity, and so we identify a new molecular pathological mechanism by which spastin mutations may cause disease.
Keywords: CHMP1B; ESCRT; IST1; endosome tubule fission; hereditary spastic paraplegia.
Copyright © 2019 Allison, Edgar and Reid.
Figures

References
Grants and funding
LinkOut - more resources
Full Text Sources