

Biallelic variants in EFEMP1 in a man with a pronounced connective tissue phenotype
- PMID: 31792352
- PMCID: PMC7080811
- DOI: 10.1038/s41431-019-0546-7
Biallelic variants in EFEMP1 in a man with a pronounced connective tissue phenotype
Abstract
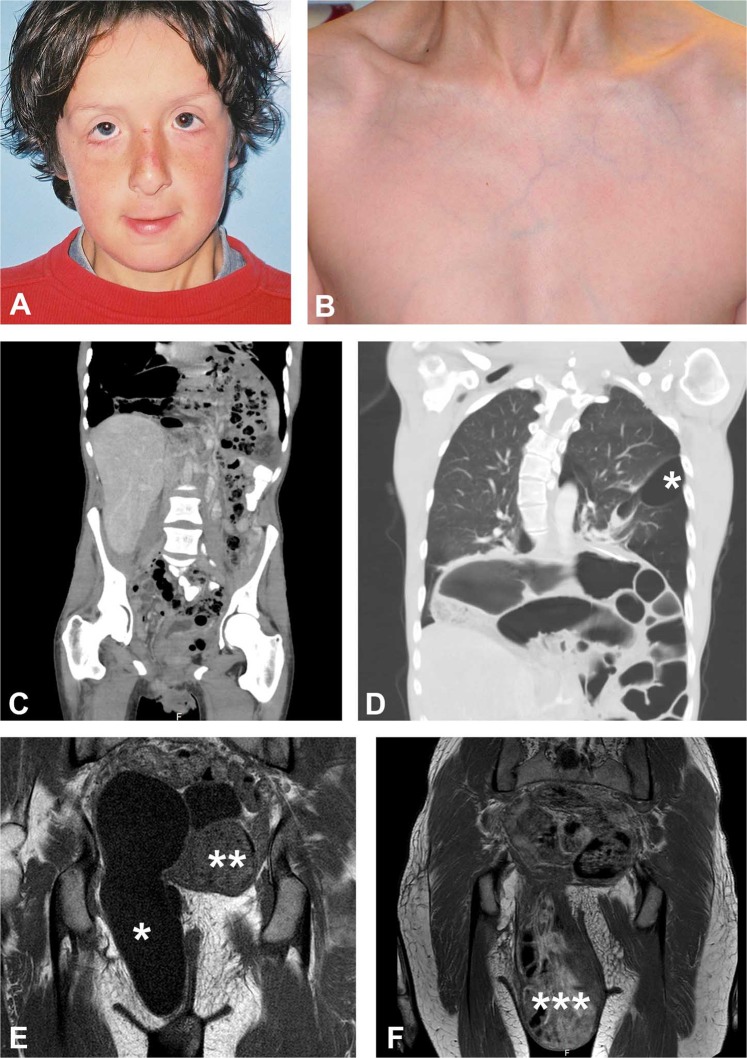
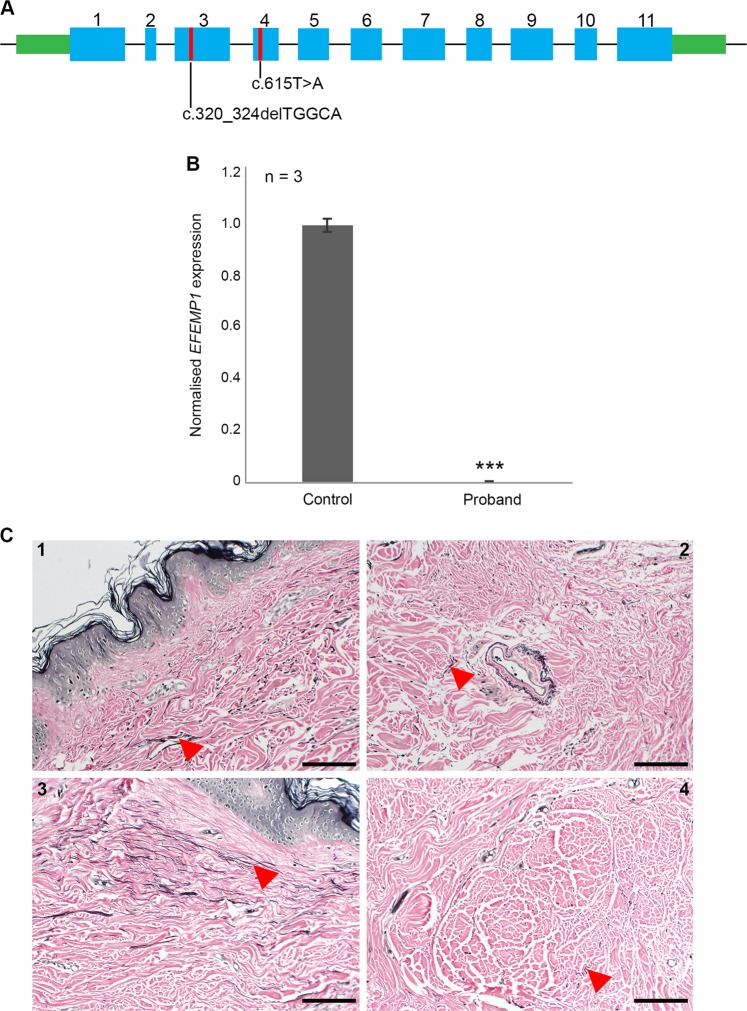
Connective tissue disorders are a spectrum of diseases that affect the integrity of tissues including skin, vasculature, and joints. They are often caused by variants that disrupt genes encoding components of extracellular matrix (ECM). The fibulin glycoproteins are ECM proteins important for integrity of tissues including dermis, retina, fascia, and vasculature. The fibulin family consists of seven members (fibulins-1 to -7) and is defined by a fibulin-type domain at the C-terminus. The family is associated with human diseases, for instance a variant in FBLN1, encoding fibulin-1, is associated with synpolydactyly, while one in EFEMP1, encoding fibulin-3, causes Doyne honeycomb degeneration of the retina. Loss-of-function of fibulins-4 and -5 causes cutis laxa, while variants in fibulins-5 and -6 are associated with age-related macular degeneration. Of note, EFEMP1 is not currently associated with any connective tissue disorder. Here we show biallelic loss-of-function variants in EFEMP1 in an individual with multiple and recurrent abdominal and thoracic herniae, myopia, hypermobile joints, scoliosis, and thin translucent skin. Fibroblasts from this individual express significantly lower EFEMP1 transcript than age-matched control cells. A skin biopsy, visualised using light microscopy, showed normal structure and abundance of elastic fibres. The phenotype of this individual is remarkably similar to the Efemp1 knockout mouse model that displays multiple herniae with premature aging and scoliosis. We conclude that loss of EFEMP1 function in this individual is the cause of a connective tissue disorder with a novel combination of phenotypic features, and can perhaps explain similar, previously reported cases in the literature.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
