

Deciphering eukaryotic gene-regulatory logic with 100 million random promoters
- PMID: 31792407
- PMCID: PMC6954276
- DOI: 10.1038/s41587-019-0315-8
Deciphering eukaryotic gene-regulatory logic with 100 million random promoters
Erratum in
-
Author Correction: Deciphering eukaryotic gene-regulatory logic with 100 million random promoters.Nat Biotechnol. 2020 Oct;38(10):1211. doi: 10.1038/s41587-020-0665-2. Nat Biotechnol. 2020. PMID: 32792646
Abstract
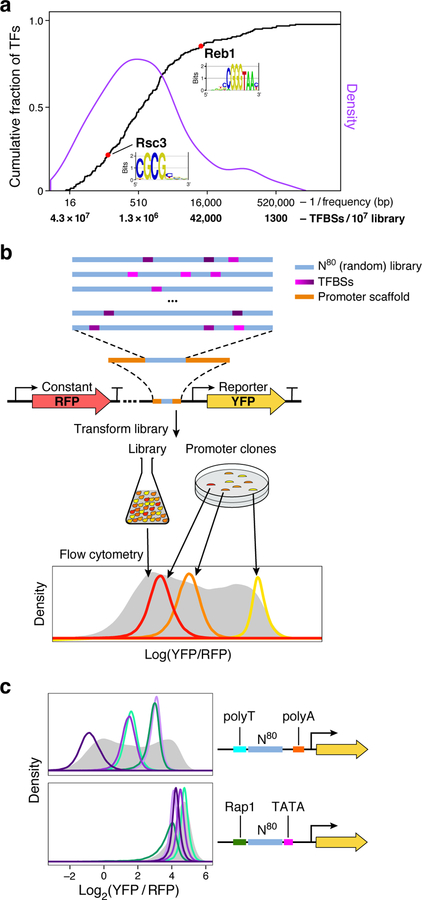
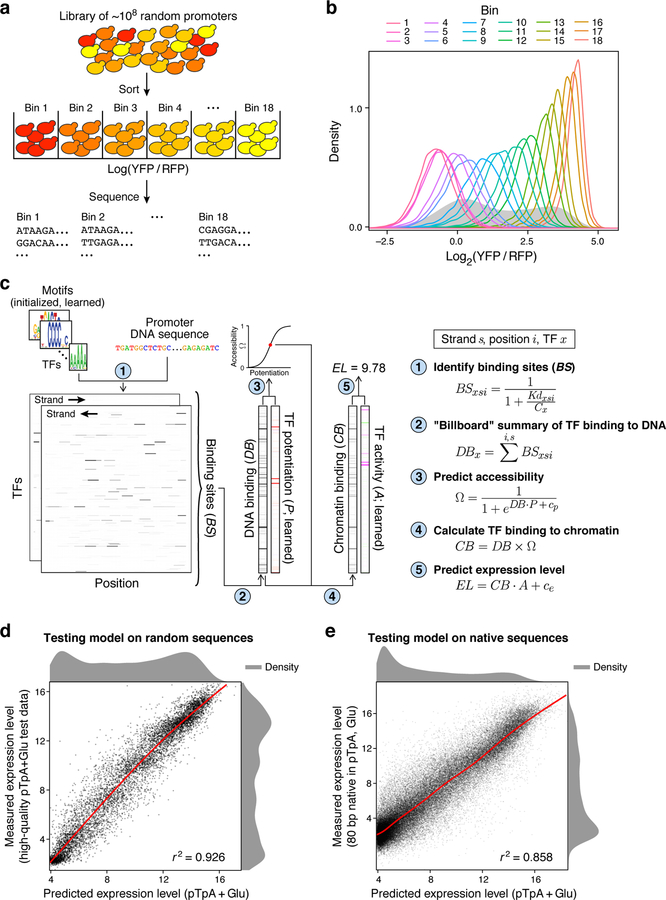
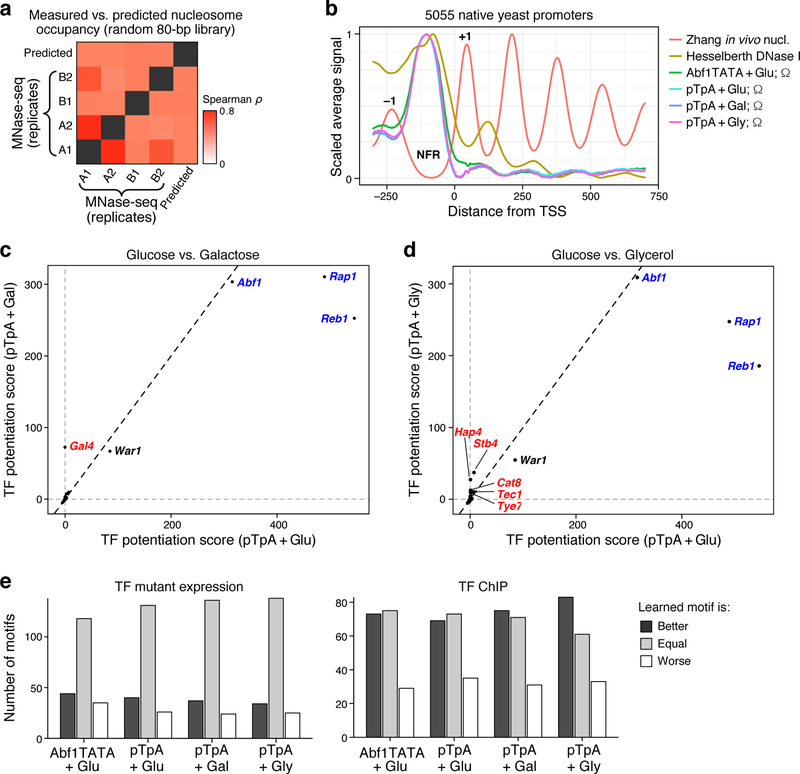
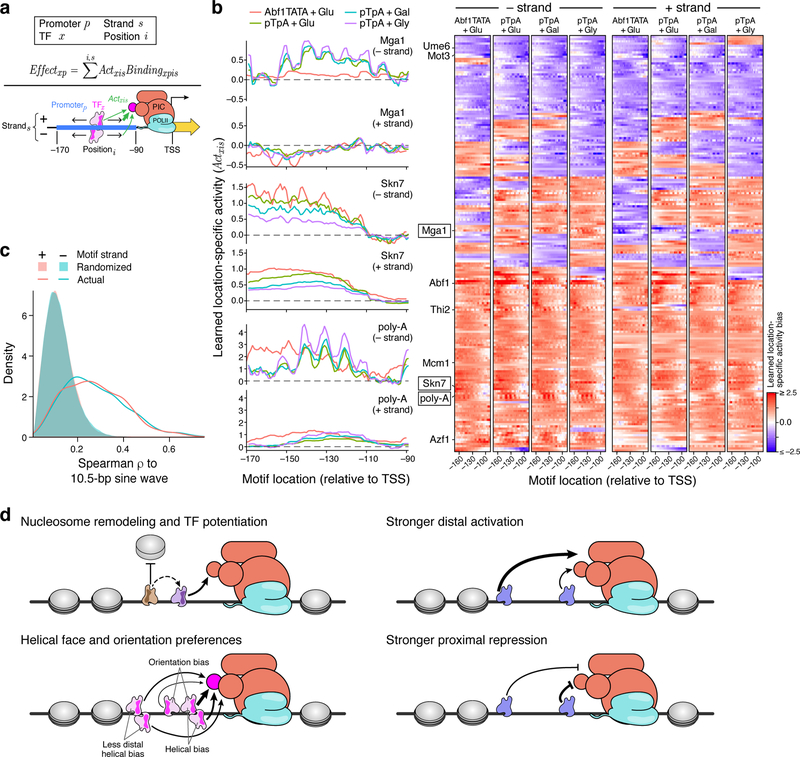
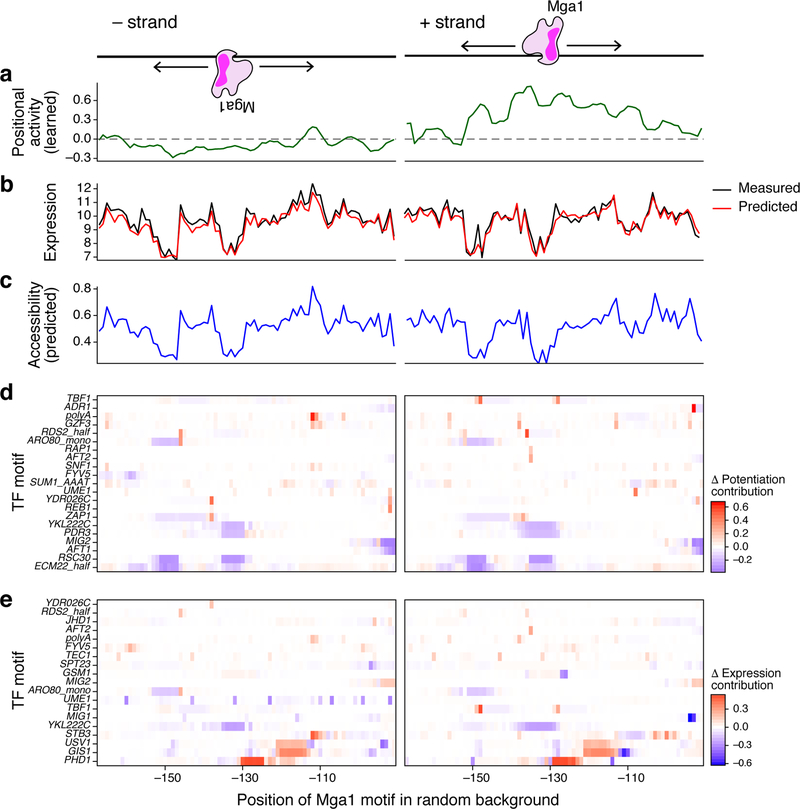
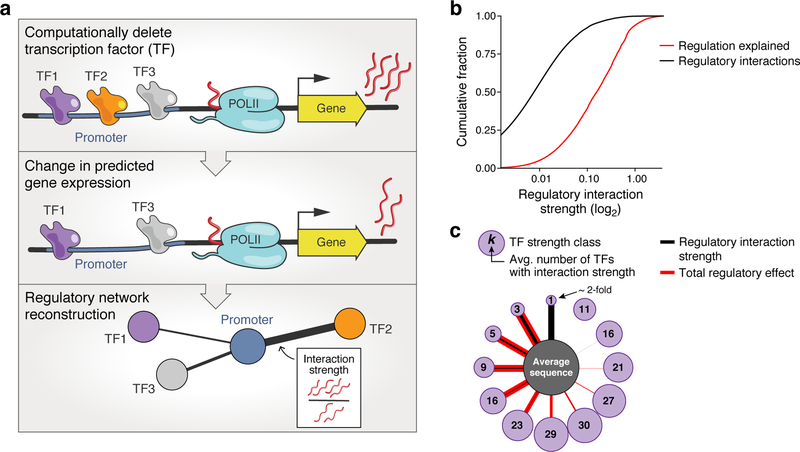
How transcription factors (TFs) interpret cis-regulatory DNA sequence to control gene expression remains unclear, largely because past studies using native and engineered sequences had insufficient scale. Here, we measure the expression output of >100 million synthetic yeast promoter sequences that are fully random. These sequences yield diverse, reproducible expression levels that can be explained by their chance inclusion of functional TF binding sites. We use machine learning to build interpretable models of transcriptional regulation that predict ~94% of the expression driven from independent test promoters and ~89% of the expression driven from native yeast promoter fragments. These models allow us to characterize each TF's specificity, activity and interactions with chromatin. TF activity depends on binding-site strand, position, DNA helical face and chromatin context. Notably, expression level is influenced by weak regulatory interactions, which confound designed-sequence studies. Our analyses show that massive-throughput assays of fully random DNA can provide the big data necessary to develop complex, predictive models of gene regulation.
Conflict of interest statement
Declaration of Interests
AR is an SAB member of ThermoFisher Scientific, Neogene Therapeutics, and Syros Pharmaceuticals and a founder of and equity holder in Celsius Therapeutics. All other authors declare no competing interests.
Figures
References
-
- Beer MA & Tavazoie S Predicting gene expression from sequence. Cell 117, 185–198 (2004). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
