

Single-cell multiomic analysis identifies regulatory programs in mixed-phenotype acute leukemia
- PMID: 31792411
- PMCID: PMC7258684
- DOI: 10.1038/s41587-019-0332-7
Single-cell multiomic analysis identifies regulatory programs in mixed-phenotype acute leukemia
Abstract
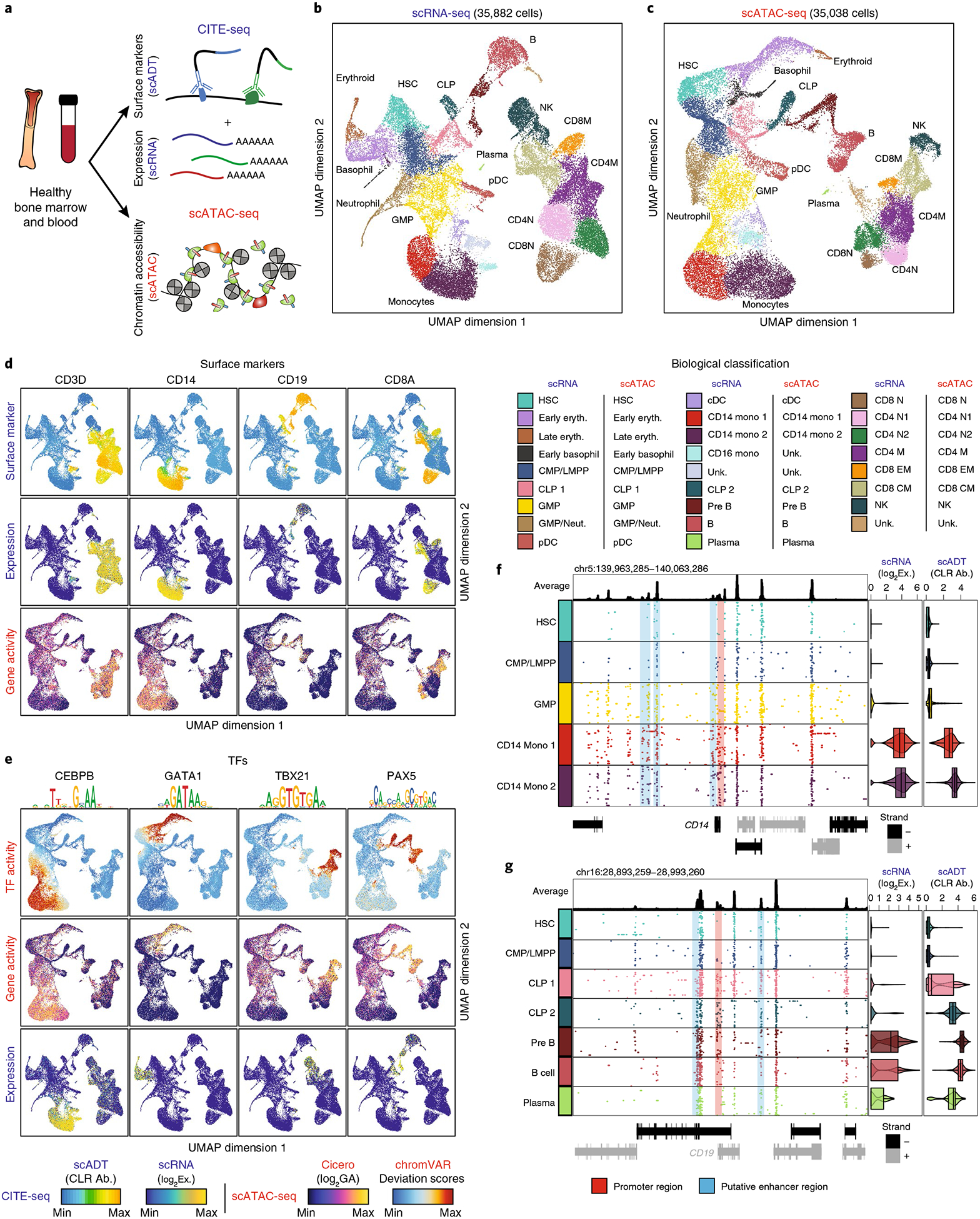
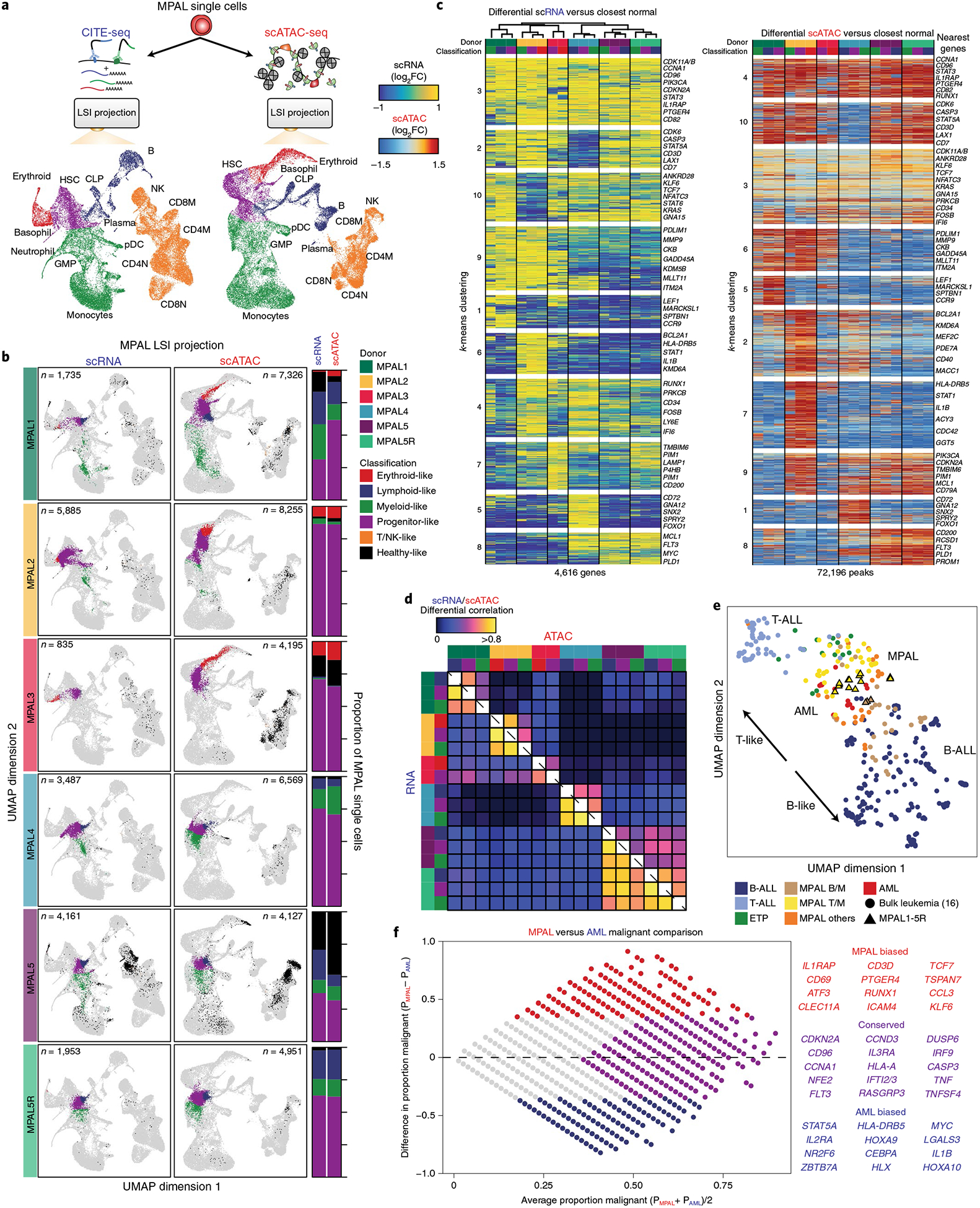
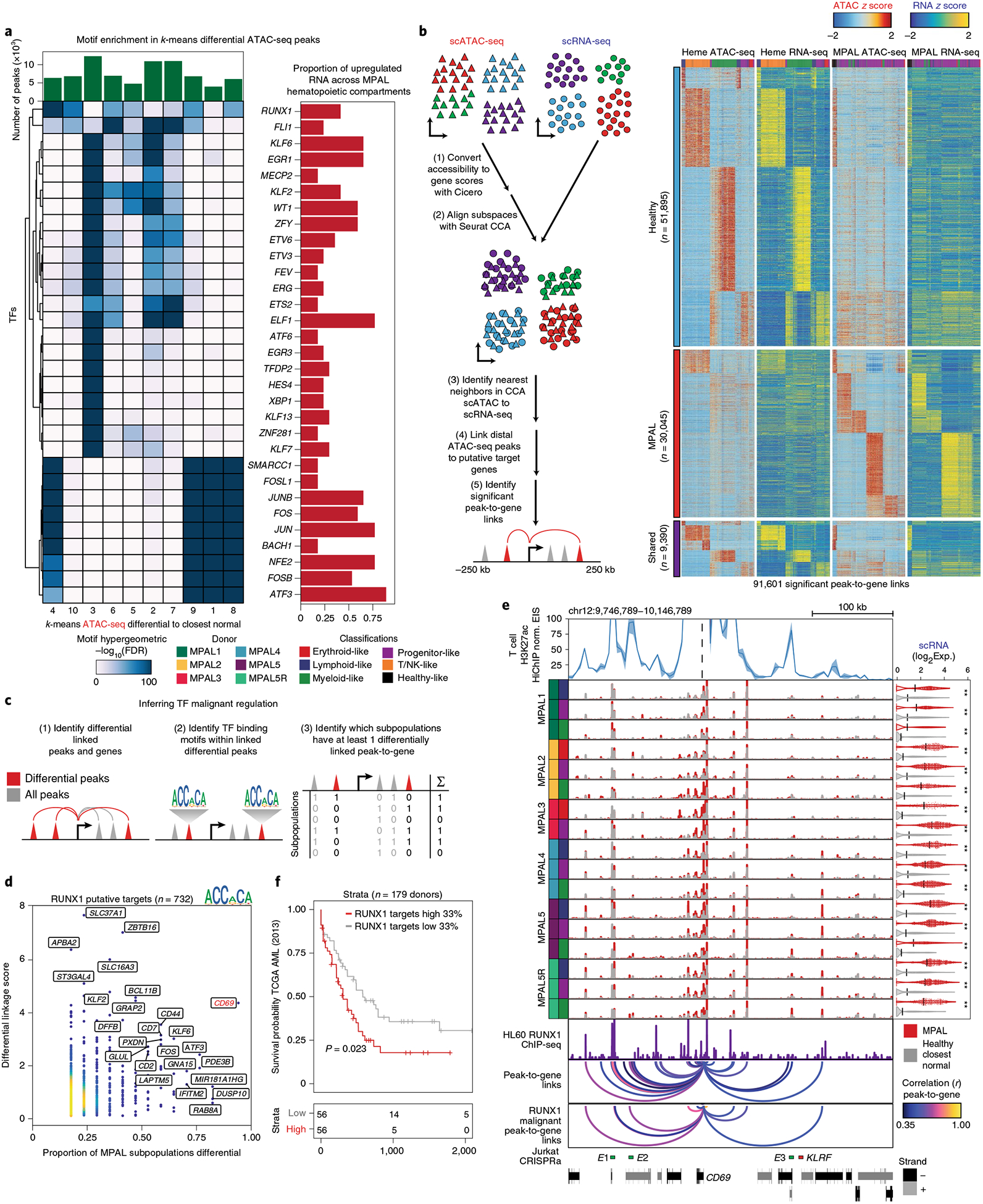
Identifying the causes of human diseases requires deconvolution of abnormal molecular phenotypes spanning DNA accessibility, gene expression and protein abundance1-3. We present a single-cell framework that integrates highly multiplexed protein quantification, transcriptome profiling and analysis of chromatin accessibility. Using this approach, we establish a normal epigenetic baseline for healthy blood development, which we then use to deconvolve aberrant molecular features within blood from patients with mixed-phenotype acute leukemia4,5. Despite widespread epigenetic heterogeneity within the patient cohort, we observe common malignant signatures across patients as well as patient-specific regulatory features that are shared across phenotypic compartments of individual patients. Integrative analysis of transcriptomic and chromatin-accessibility maps identified 91,601 putative peak-to-gene linkages and transcription factors that regulate leukemia-specific genes, such as RUNX1-linked regulatory elements proximal to the marker gene CD69. These results demonstrate how integrative, multiomic analysis of single cells within the framework of normal development can reveal both distinct and shared molecular mechanisms of disease from patient samples.
Conflict of interest statement
Competing interests
R.M. is a founder of, is an equity holder in, and serves on the board of directors of Forty Seven. H.Y.C. has affiliations with Accent Therapeutics (founder and scientific advisory board (SAB) member), 10x Genomics (SAB member), Boundless Bio (cofounder, SAB), Arsenal Biosciences (SAB) and Spring Discovery (SAB member). W.J.G. has affiliations with 10x Genomics (consultant), Guardant Health (consultant) and Protillion Biosciences (co-founder and consultant).
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
