

ASC-1 Is a Cell Cycle Regulator Associated with Severe and Mild Forms of Myopathy
- PMID: 31794073
- PMCID: PMC6980348
- DOI: 10.1002/ana.25660
ASC-1 Is a Cell Cycle Regulator Associated with Severe and Mild Forms of Myopathy
Abstract
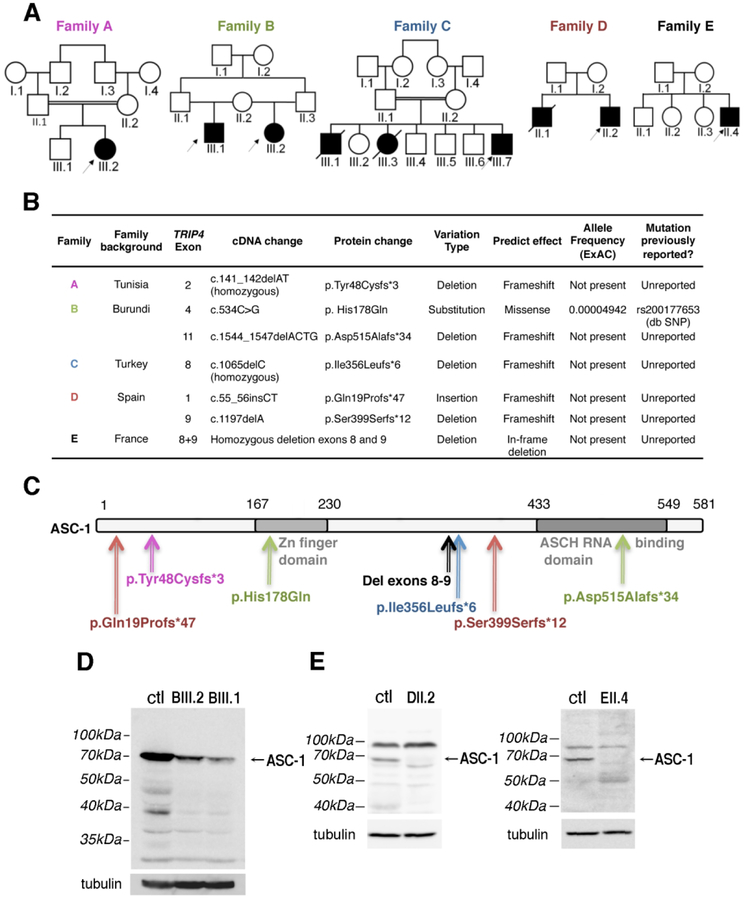
Objective: Recently, the ASC-1 complex has been identified as a mechanistic link between amyotrophic lateral sclerosis and spinal muscular atrophy (SMA), and 3 mutations of the ASC-1 gene TRIP4 have been associated with SMA or congenital myopathy. Our goal was to define ASC-1 neuromuscular function and the phenotypical spectrum associated with TRIP4 mutations.
Methods: Clinical, molecular, histological, and magnetic resonance imaging studies were made in 5 families with 7 novel TRIP4 mutations. Fluorescence activated cell sorting and Western blot were performed in patient-derived fibroblasts and muscles and in Trip4 knocked-down C2C12 cells.
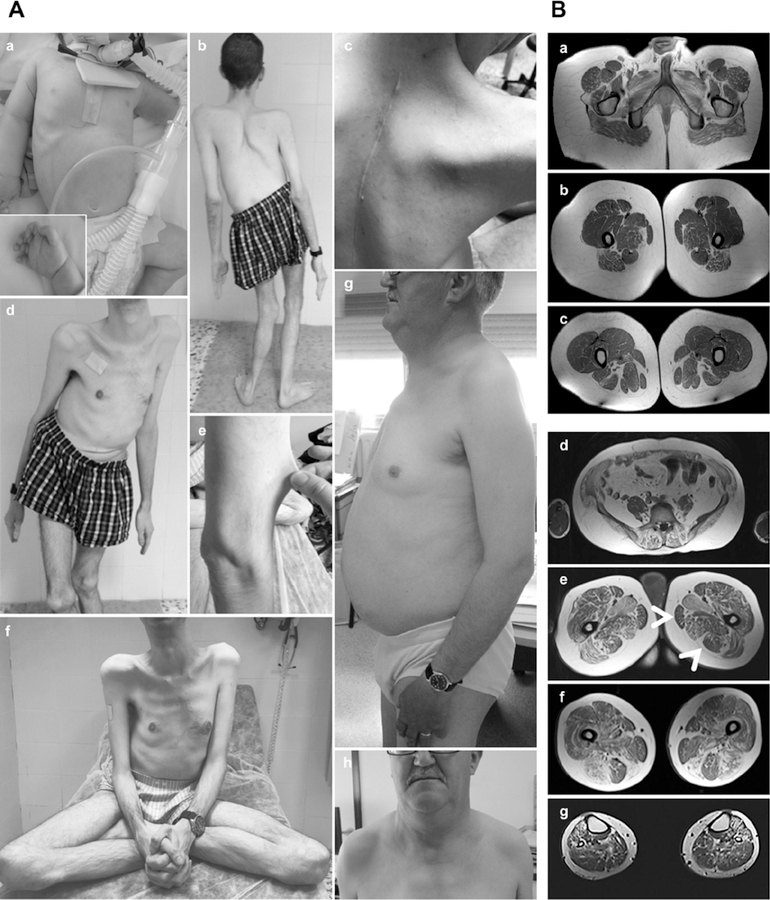
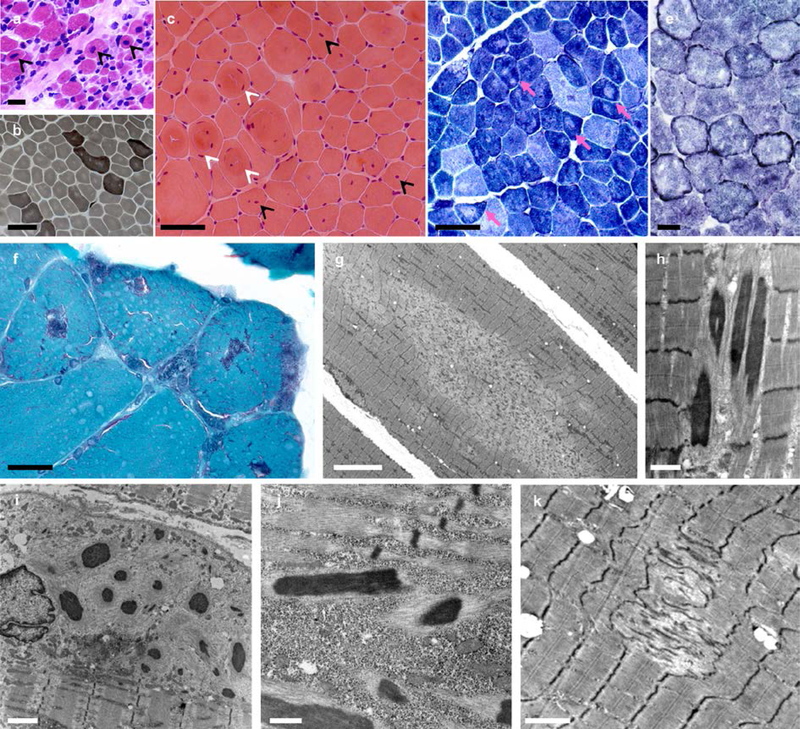
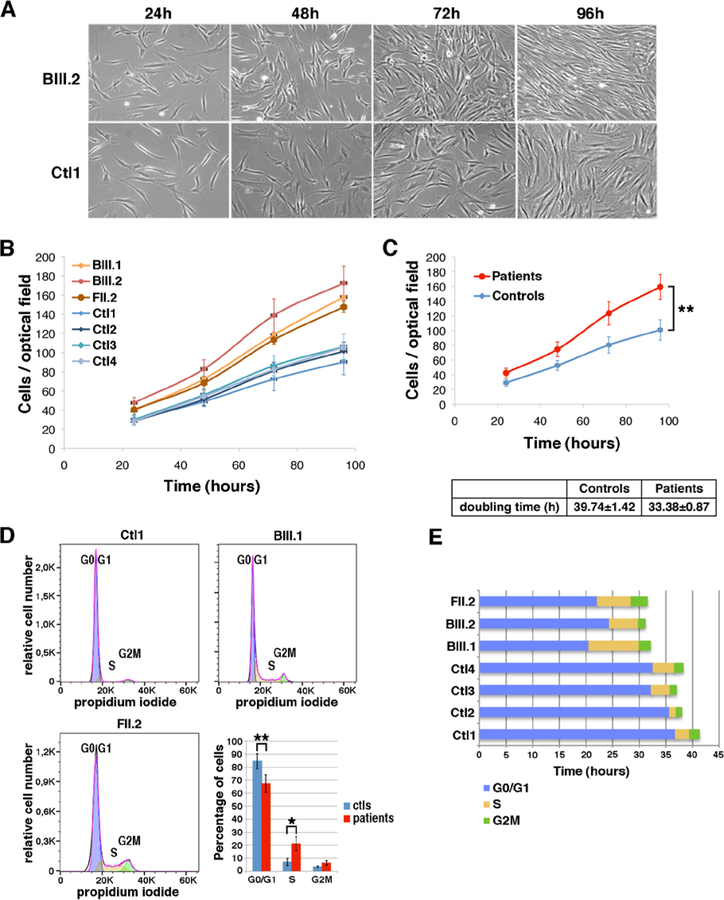
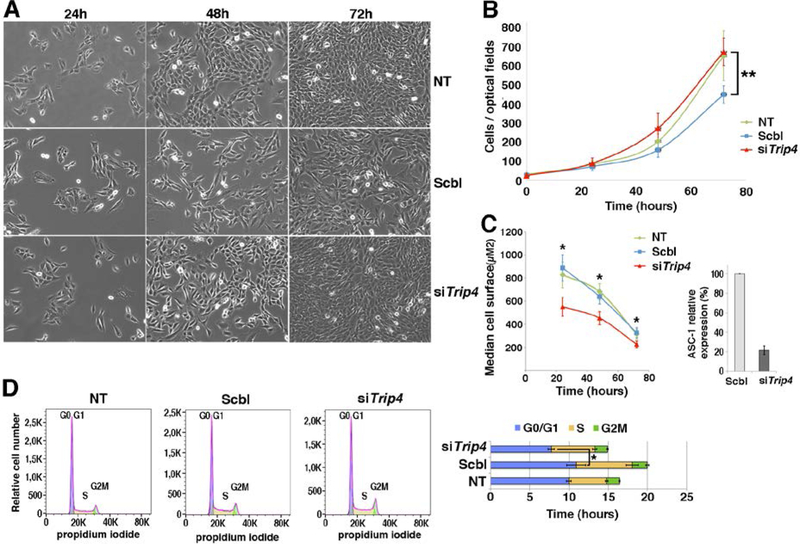
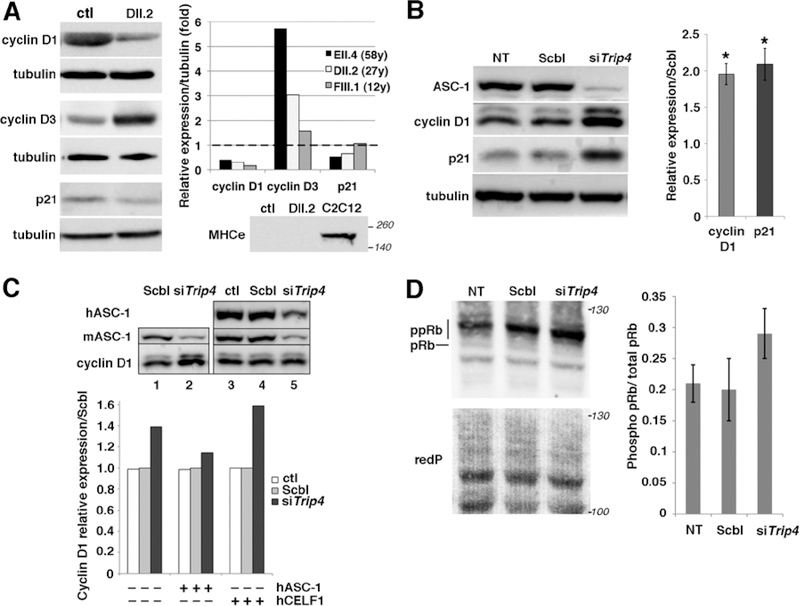
Results: All mutations caused ASC-1 protein depletion. The clinical phenotype was purely myopathic, ranging from lethal neonatal to mild ambulatory adult patients. It included early onset axial and proximal weakness, scoliosis, rigid spine, dysmorphic facies, cutaneous involvement, respiratory failure, and in the older cases, dilated cardiomyopathy. Muscle biopsies showed multiminicores, nemaline rods, cytoplasmic bodies, caps, central nuclei, rimmed fibers, and/or mild endomysial fibrosis. ASC-1 depletion in C2C12 and in patient-derived fibroblasts and muscles caused accelerated proliferation, altered expression of cell cycle proteins, and/or shortening of the G0/G1 cell cycle phase leading to cell size reduction.
Interpretation: Our results expand the phenotypical and molecular spectrum of TRIP4-associated disease to include mild adult forms with or without cardiomyopathy, associate ASC-1 depletion with isolated primary muscle involvement, and establish TRIP4 as a causative gene for several congenital muscle diseases, including nemaline, core, centronuclear, and cytoplasmic-body myopathies. They also identify ASC-1 as a novel cell cycle regulator with a key role in cell proliferation, and underline transcriptional coregulation defects as a novel pathophysiological mechanism. ANN NEUROL 2020;87:217-232.
© 2019 American Neurological Association.
Conflict of interest statement
POTENTIAL CONFLICTS OF INTEREST
The authors declare that they have no conflict of interest.
Figures
References
-
- Schorling D, Kirschner J, Bönnemann C. Congenital Muscular Dystrophies and Myopathies: An Overview and Update [Internet]. Neuropediatrics 2017;48(4):247–261. - PubMed
-
- Jungbluth H, Ochala J, Treves S, Gautel M. Current and future therapeutic approaches to the congenital myopathies. Semin. Cell Dev. Biol. 2017;64:191–200. - PubMed
-
- Kaplan J-C, Hamroun D. The 2016 version of the gene table of monogenic neuromuscular disorders (nuclear genome). Neuromuscul. Disord 2015;25(12):991–1020. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- UM1 HG008900/HG/NHGRI NIH HHS/United States
- University of Paris/International
- National Institute of Health and Medical Research/International
- PI14/00738/Carlos III Institute of Health/International
- PI14/00738/Instituto de Salud Carlos III/International
- MDA602235/Muscular Dystrophy Association/International
- #20923/Association Française contre les Myopathies/International
- R01 HD075802/HD/NICHD NIH HHS/United States
- 21267/French Association against Myopathies/International
- U54 HD090255/HD/NICHD NIH HHS/United States
- R03/75-79/League against Cancer/International
- National Center for Scientific Research/International
- 20923/French Association against Myopathies/International
- European Regional Development Fund/International
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
