

The interaction of ceramide 1-phosphate with group IVA cytosolic phospholipase A2 coordinates acute wound healing and repair
- PMID: 31796632
- PMCID: PMC8103357
- DOI: 10.1126/scisignal.aav5918
The interaction of ceramide 1-phosphate with group IVA cytosolic phospholipase A2 coordinates acute wound healing and repair
Abstract
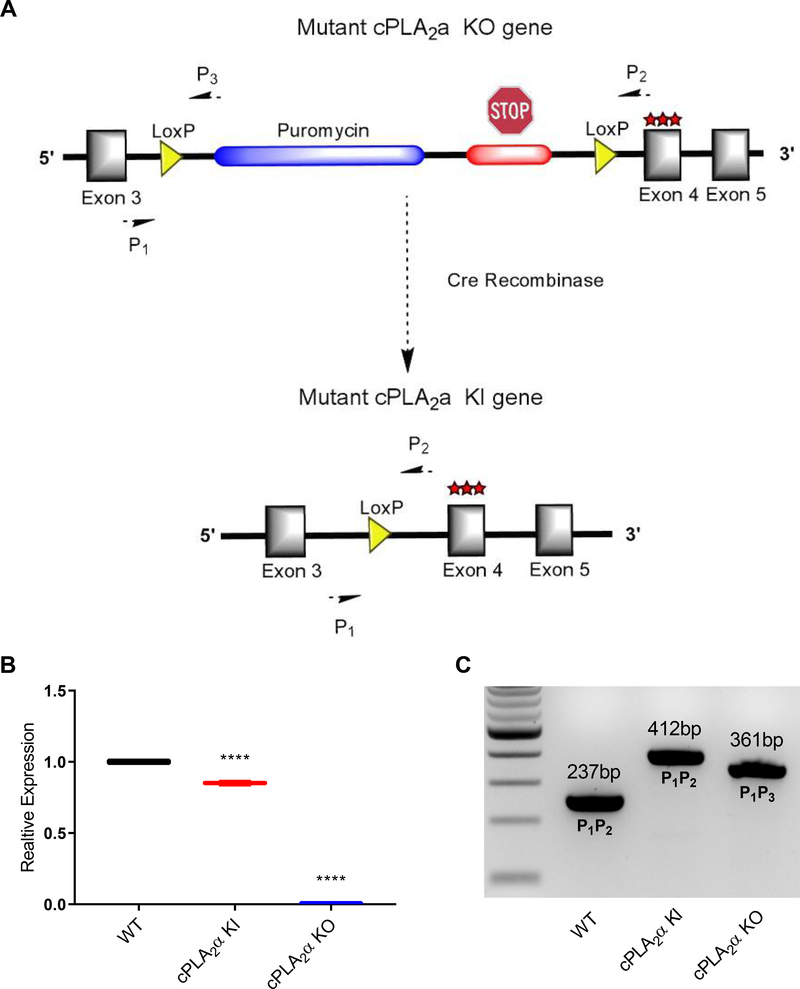
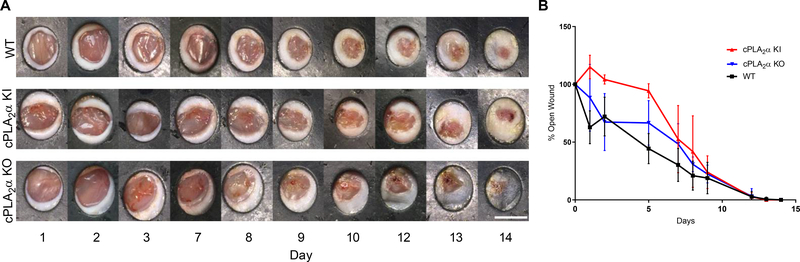
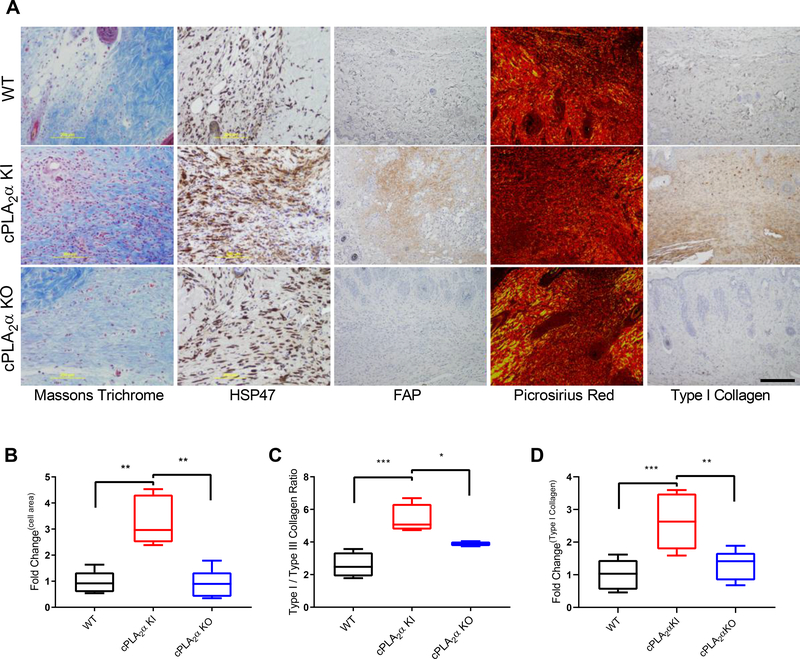
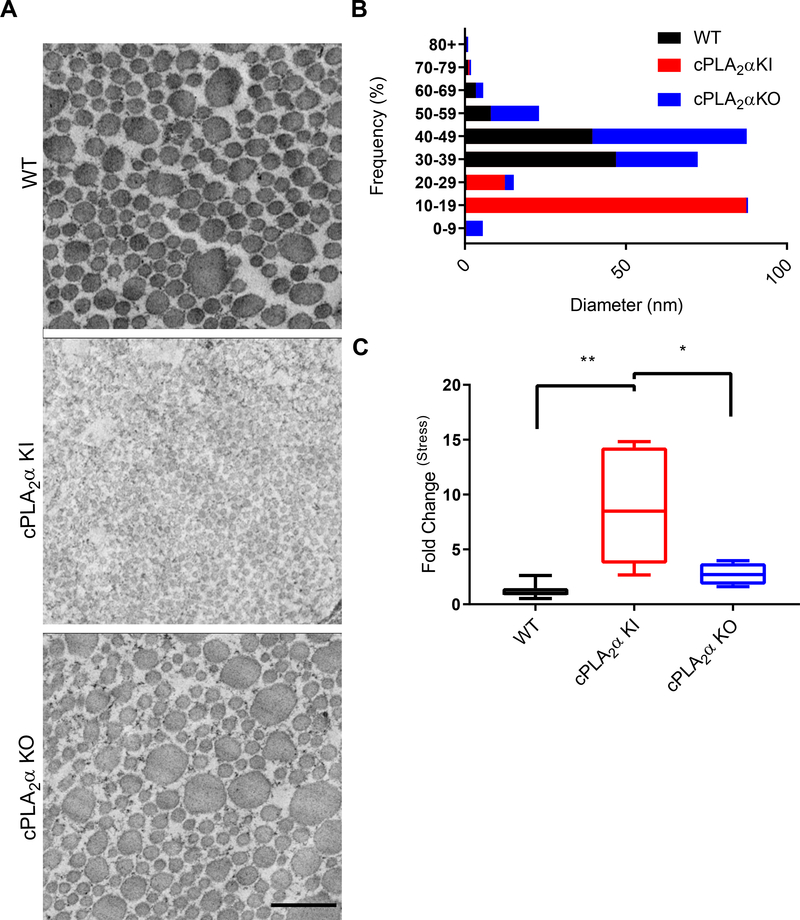
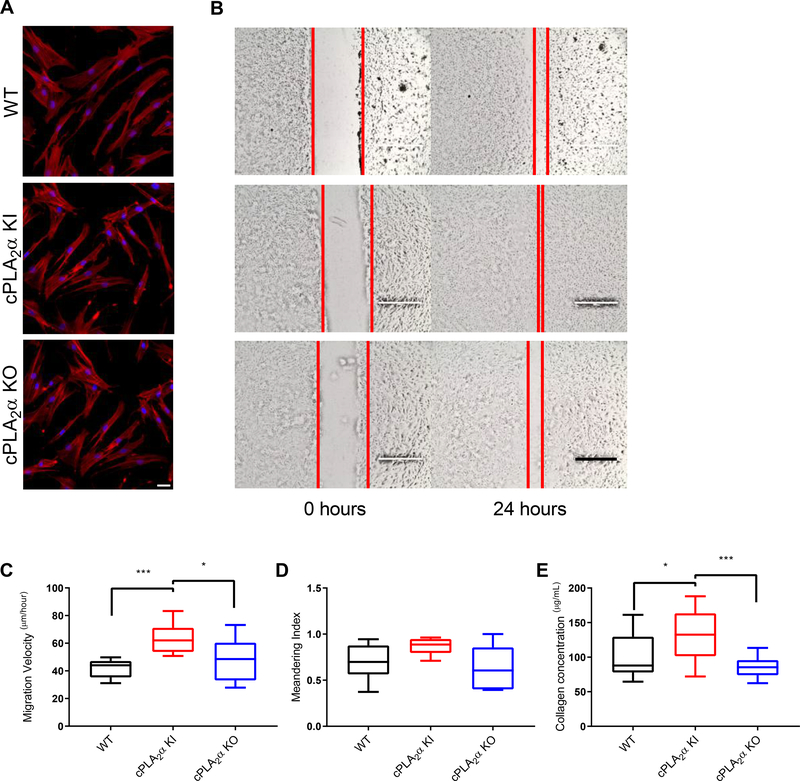
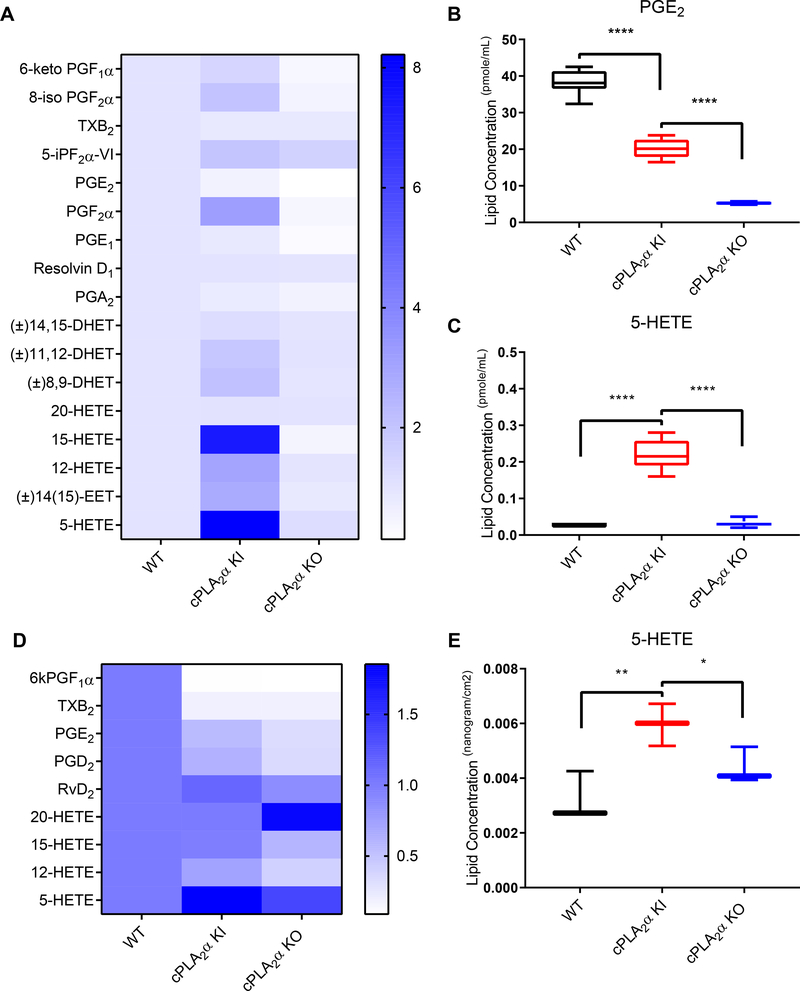
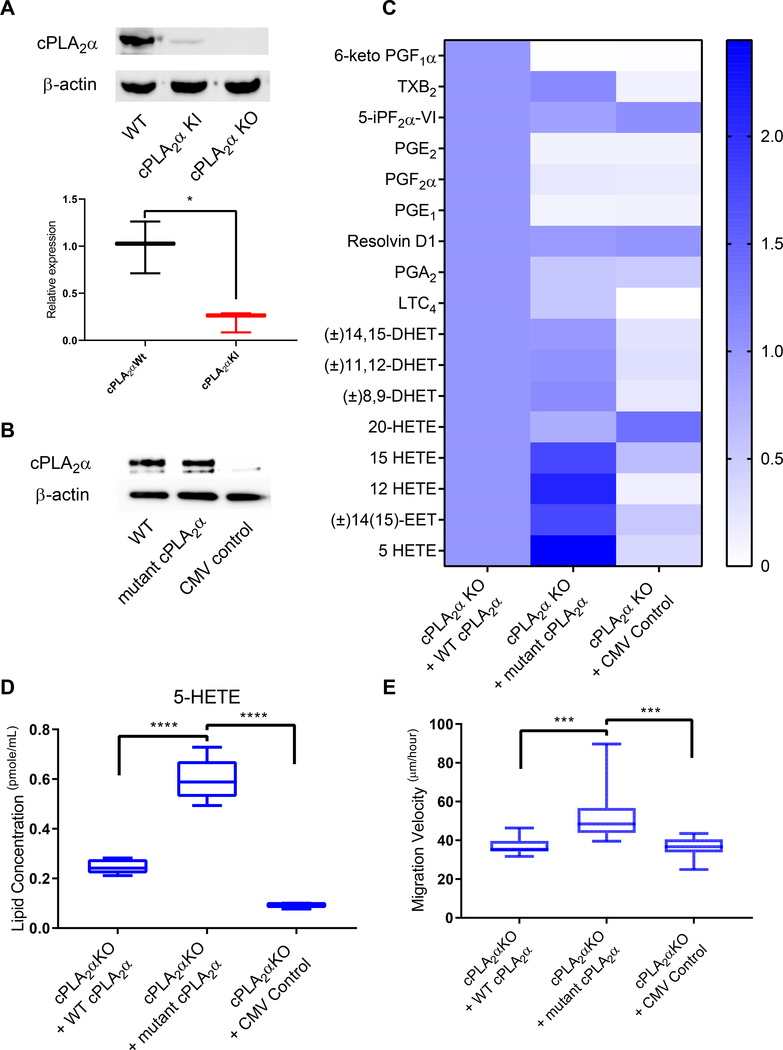
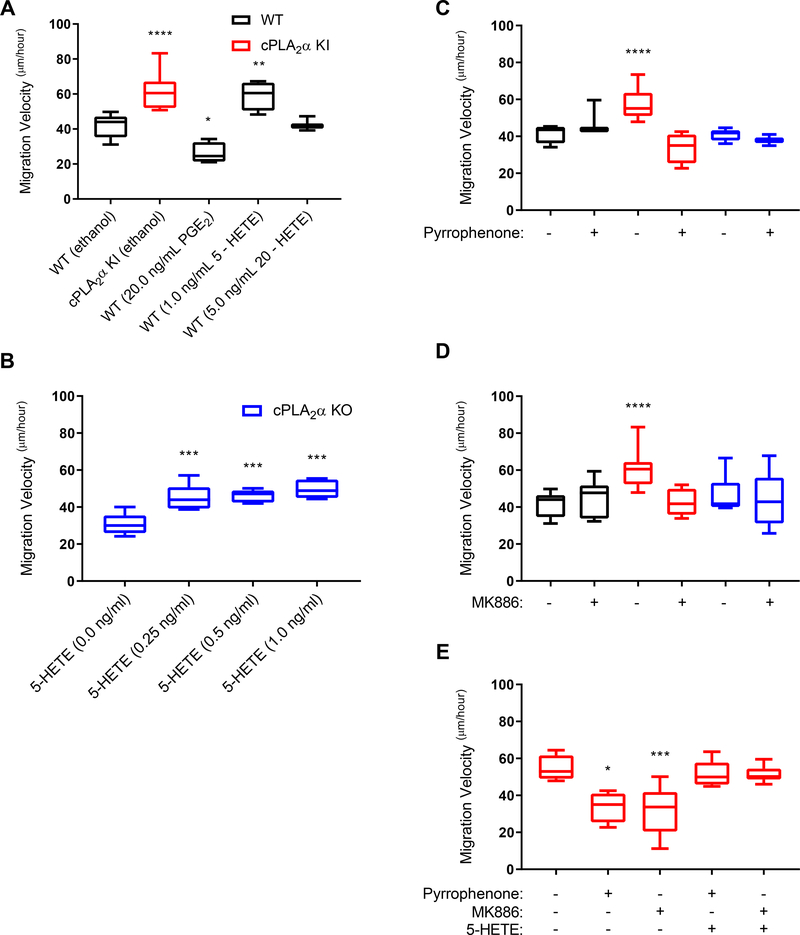
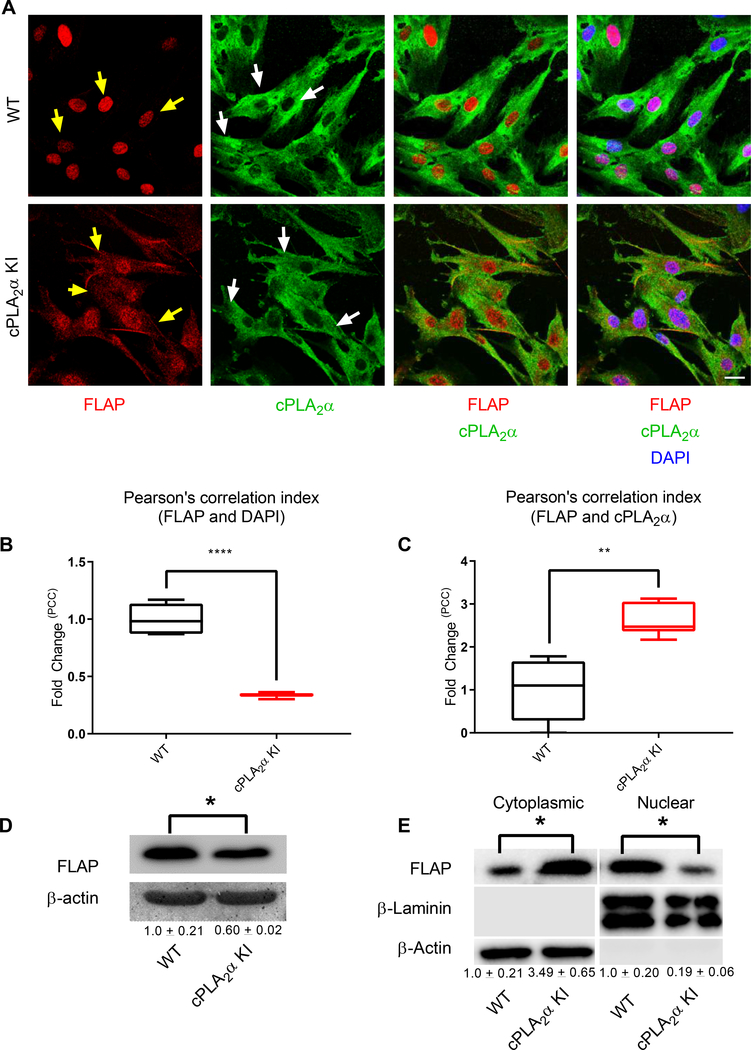
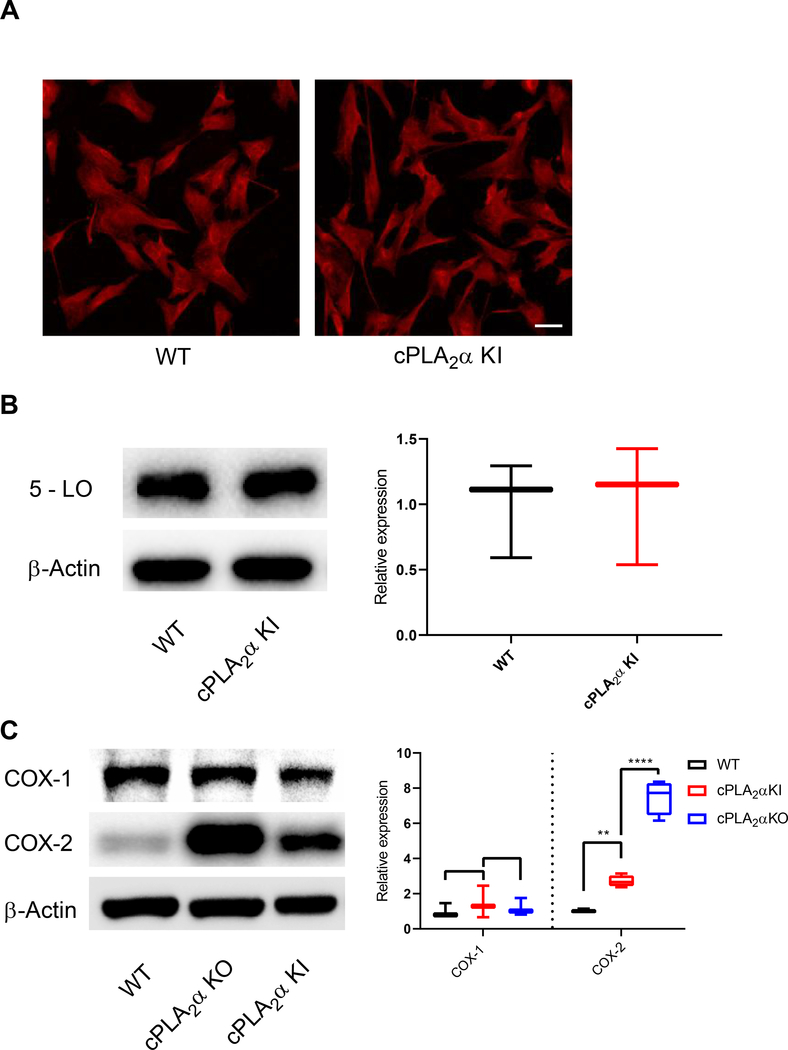
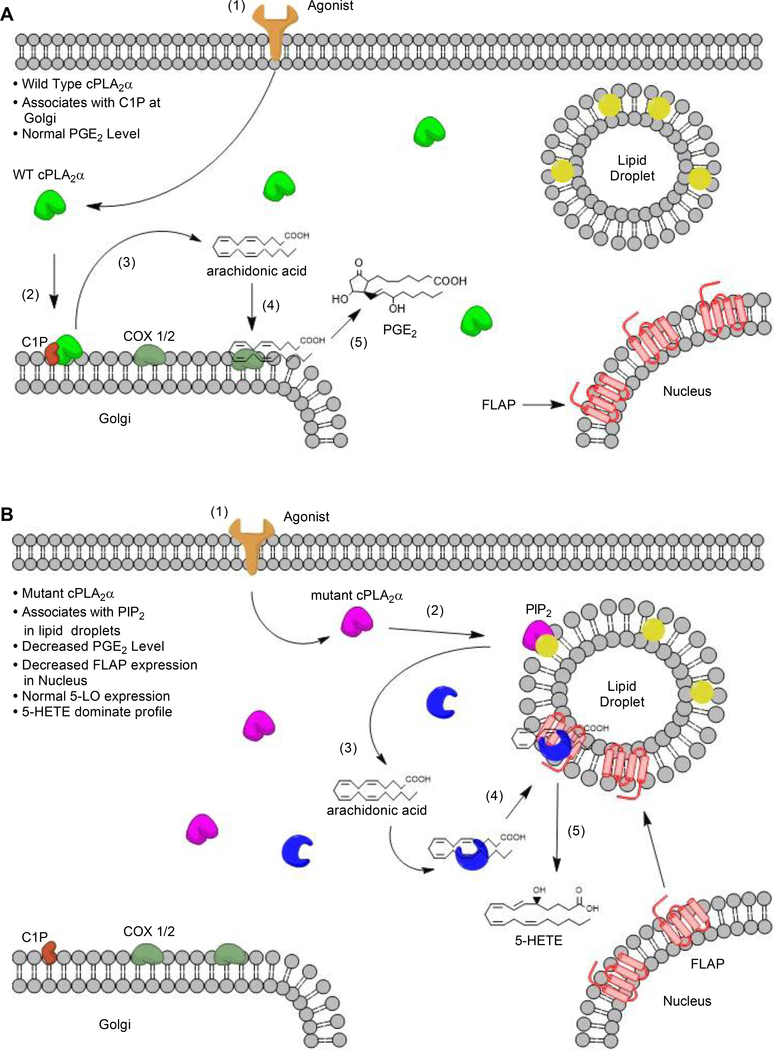
The sphingolipid ceramide 1-phosphate (C1P) directly binds to and activates group IVA cytosolic phospholipase A2 (cPLA2α) to stimulate the production of eicosanoids. Because eicosanoids are important in wound healing, we examined the repair of skin wounds in knockout (KO) mice lacking cPLA2α and in knock-in (KI) mice in which endogenous cPLA2α was replaced with a mutant form having an ablated C1P interaction site. Wound closure rate was not affected in the KO or KI mice, but wound maturation was enhanced in the KI mice compared to that in wild-type controls. Wounds in KI mice displayed increased infiltration of dermal fibroblasts into the wound environment, increased wound tensile strength, and a higher ratio of type I:type III collagen. In vitro, primary dermal fibroblasts (pDFs) from KI mice showed substantially increased collagen deposition and migration velocity compared to pDFs from wild-type and KO mice. KI mice also showed an altered eicosanoid profile of reduced proinflammatory prostaglandins (PGE2 and TXB2) and an increased abundance of certain hydroxyeicosatetraenoic acid (HETE) species. Specifically, an increase in 5-HETE enhanced dermal fibroblast migration and collagen deposition. This gain-of-function role for the mutant cPLA2α was also linked to the relocalization of cPLA2α and 5-HETE biosynthetic enzymes to the cytoplasm and cytoplasmic vesicles. These findings demonstrate the regulation of key wound-healing mechanisms in vivo by a defined protein-lipid interaction and provide insights into the roles that cPLA2α and eicosanoids play in orchestrating wound repair.
Copyright © 2019 The Authors, some rights reserved; exclusive licensee American Association for the Advancement of Science. No claim to original U.S. Government Works.
Conflict of interest statement
Figures
Similar articles
-
Ceramide kinase regulates acute wound healing by suppressing 5-oxo-ETE biosynthesis and signaling via its receptor OXER1.J Lipid Res. 2022 Apr;63(4):100187. doi: 10.1016/j.jlr.2022.100187. Epub 2022 Feb 24. J Lipid Res. 2022. PMID: 35219746 Free PMC article.
-
Skewing cPLA2α activity toward oxoeicosanoid production promotes neutrophil N2 polarization, wound healing, and the response to sepsis.Sci Signal. 2023 Jul 11;16(793):eadd6527. doi: 10.1126/scisignal.add6527. Epub 2023 Jul 11. Sci Signal. 2023. PMID: 37433004 Free PMC article.
-
Study of the role of cytosolic phospholipase A2 alpha in eicosanoid generation and thymocyte maturation in the thymus.PLoS One. 2015 May 13;10(5):e0126204. doi: 10.1371/journal.pone.0126204. eCollection 2015. PLoS One. 2015. PMID: 25969996 Free PMC article.
-
Ceramide kinase and the ceramide-1-phosphate/cPLA2alpha interaction as a therapeutic target.Curr Drug Targets. 2008 Aug;9(8):674-82. doi: 10.2174/138945008785132349. Curr Drug Targets. 2008. PMID: 18691014 Review.
-
The role of cytosolic phospholipase A2-alfa in regulation of phagocytic functions.Biochim Biophys Acta. 2006 Nov;1761(11):1323-34. doi: 10.1016/j.bbalip.2006.09.004. Epub 2006 Sep 15. Biochim Biophys Acta. 2006. PMID: 17046321 Review.
Cited by
-
Fluoxetine restrains allergic inflammation by targeting an FcɛRI-ATP positive feedback loop in mast cells.Sci Signal. 2023 Sep 12;16(802):eabc9089. doi: 10.1126/scisignal.abc9089. Epub 2023 Sep 12. Sci Signal. 2023. PMID: 37699080 Free PMC article.
-
Oral Supplementation of n-3 Polyunsaturated Fatty Acids (n-3-PUFA) Can Prevent TBI-Induced Visual, Motor, and Emotional Deficits in Mice.Mol Neurobiol. 2025 Sep;62(9):11972-11985. doi: 10.1007/s12035-025-05019-9. Epub 2025 May 9. Mol Neurobiol. 2025. PMID: 40346443 Free PMC article.
-
RNA splicing variants of the novel long non-coding RNA, CyKILR, possess divergent biological functions in non-small cell lung cancer.Mol Ther Nucleic Acids. 2024 Dec 5;36(1):102412. doi: 10.1016/j.omtn.2024.102412. eCollection 2025 Mar 11. Mol Ther Nucleic Acids. 2024. PMID: 39807365 Free PMC article.
-
Ceramide-1-phosphate is a regulator of Golgi structure and is co-opted by the obligate intracellular bacterial pathogen Anaplasma phagocytophilum.mBio. 2024 Apr 10;15(4):e0029924. doi: 10.1128/mbio.00299-24. Epub 2024 Feb 28. mBio. 2024. PMID: 38415594 Free PMC article.
-
Ceramide Kinase Inhibition Drives Ferroptosis and Sensitivity to Cisplatin in Mutant KRAS Lung Cancer by Dysregulating VDAC-Mediated Mitochondria Function.Mol Cancer Res. 2022 Sep 2;20(9):1429-1442. doi: 10.1158/1541-7786.MCR-22-0085. Mol Cancer Res. 2022. PMID: 35560154 Free PMC article.
References
-
- Enoch S, and Price P. 2004. Pathophysiology-Of-Healing @ www.worldwidewounds.com..
-
- Clark RAF 1988. Wound Repair. In The Molecular and Cellular Biology of Wound Repair Springer US, Boston, MA. 3–50.
-
- Versteeg HH, Heemskerk JWM, Levi M, and Reitsma PH. 2013. New Fundamentals in Hemostasis. Physiol. Rev 93: 327–358. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
