

An independent poor-prognosis subtype of breast cancer defined by a distinct tumor immune microenvironment
- PMID: 31796750
- PMCID: PMC6890706
- DOI: 10.1038/s41467-019-13329-5
An independent poor-prognosis subtype of breast cancer defined by a distinct tumor immune microenvironment
Abstract
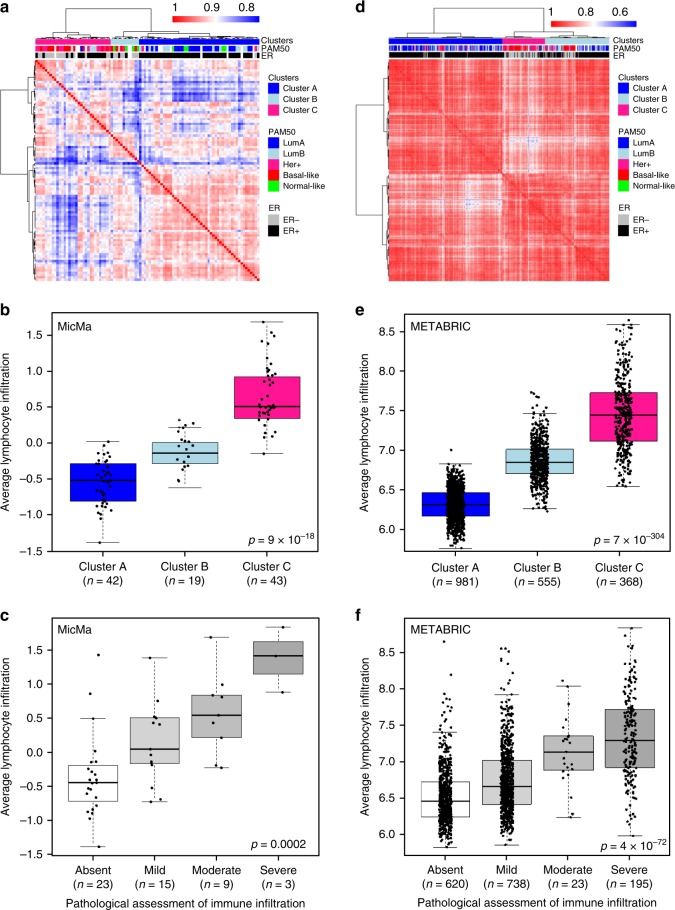
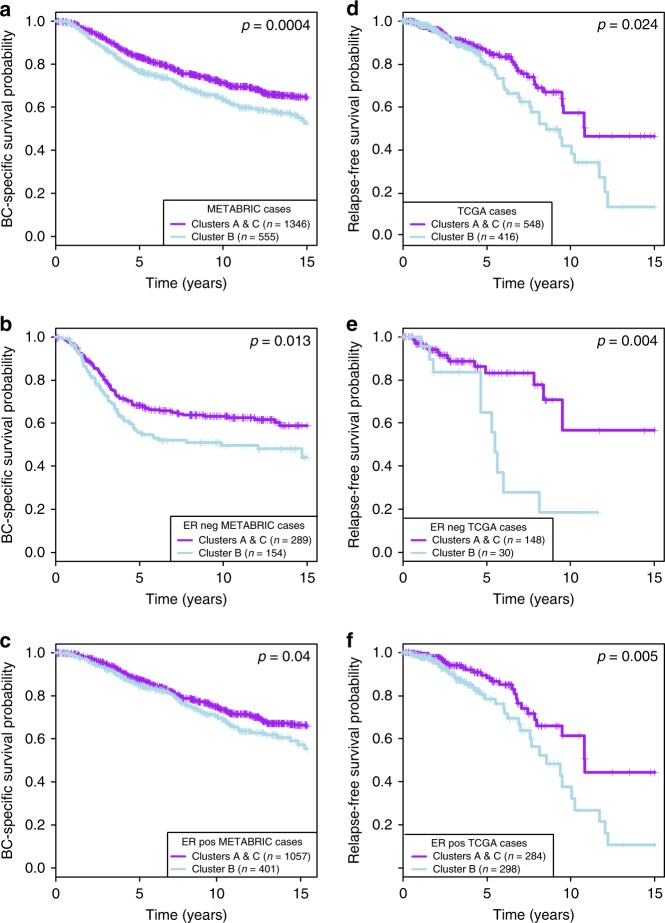
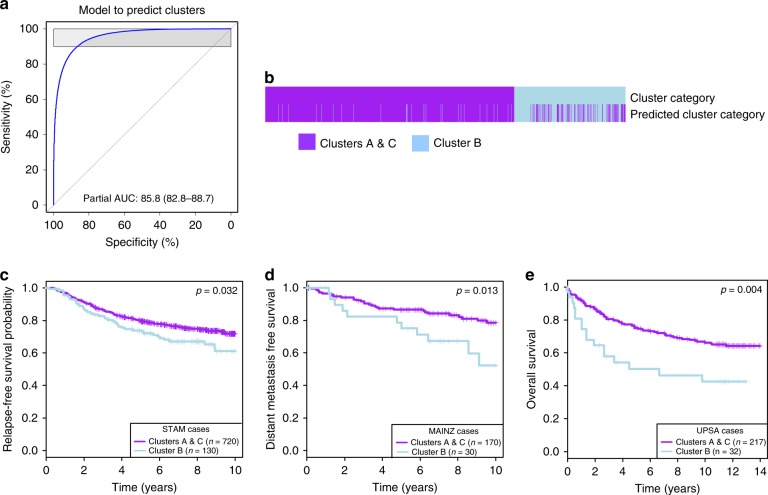
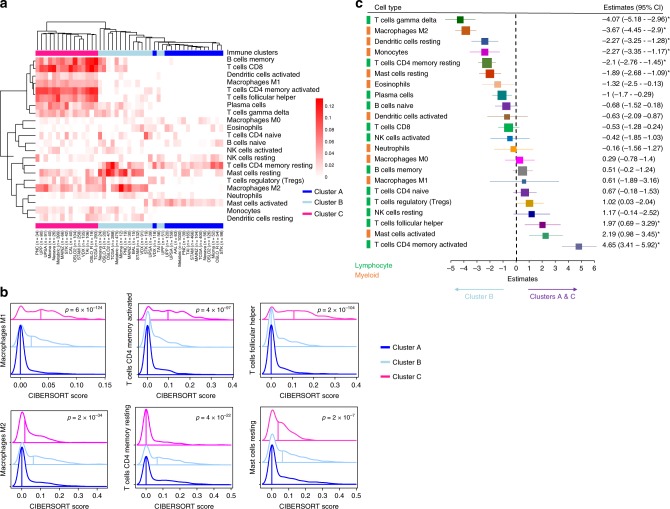
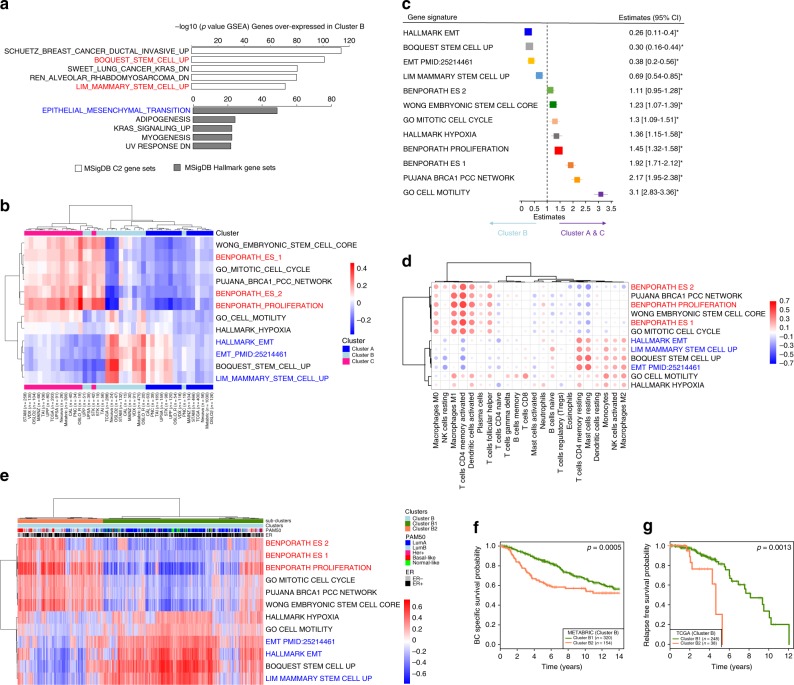
How mixtures of immune cells associate with cancer cell phenotype and affect pathogenesis is still unclear. In 15 breast cancer gene expression datasets, we invariably identify three clusters of patients with gradual levels of immune infiltration. The intermediate immune infiltration cluster (Cluster B) is associated with a worse prognosis independently of known clinicopathological features. Furthermore, immune clusters are associated with response to neoadjuvant chemotherapy. In silico dissection of the immune contexture of the clusters identified Cluster A as immune cold, Cluster C as immune hot while Cluster B has a pro-tumorigenic immune infiltration. Through phenotypical analysis, we find epithelial mesenchymal transition and proliferation associated with the immune clusters and mutually exclusive in breast cancers. Here, we describe immune clusters which improve the prognostic accuracy of immune contexture in breast cancer. Our discovery of a novel independent prognostic factor in breast cancer highlights a correlation between tumor phenotype and immune contexture.
Conflict of interest statement
The authors declare no competing interests.
Figures

References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
