

Visualizing structure and transitions in high-dimensional biological data
- PMID: 31796933
- PMCID: PMC7073148
- DOI: 10.1038/s41587-019-0336-3
Visualizing structure and transitions in high-dimensional biological data
Erratum in
-
Author Correction: Visualizing structure and transitions in high-dimensional biological data.Nat Biotechnol. 2020 Jan;38(1):108. doi: 10.1038/s41587-019-0395-5. Nat Biotechnol. 2020. PMID: 31896828
Abstract
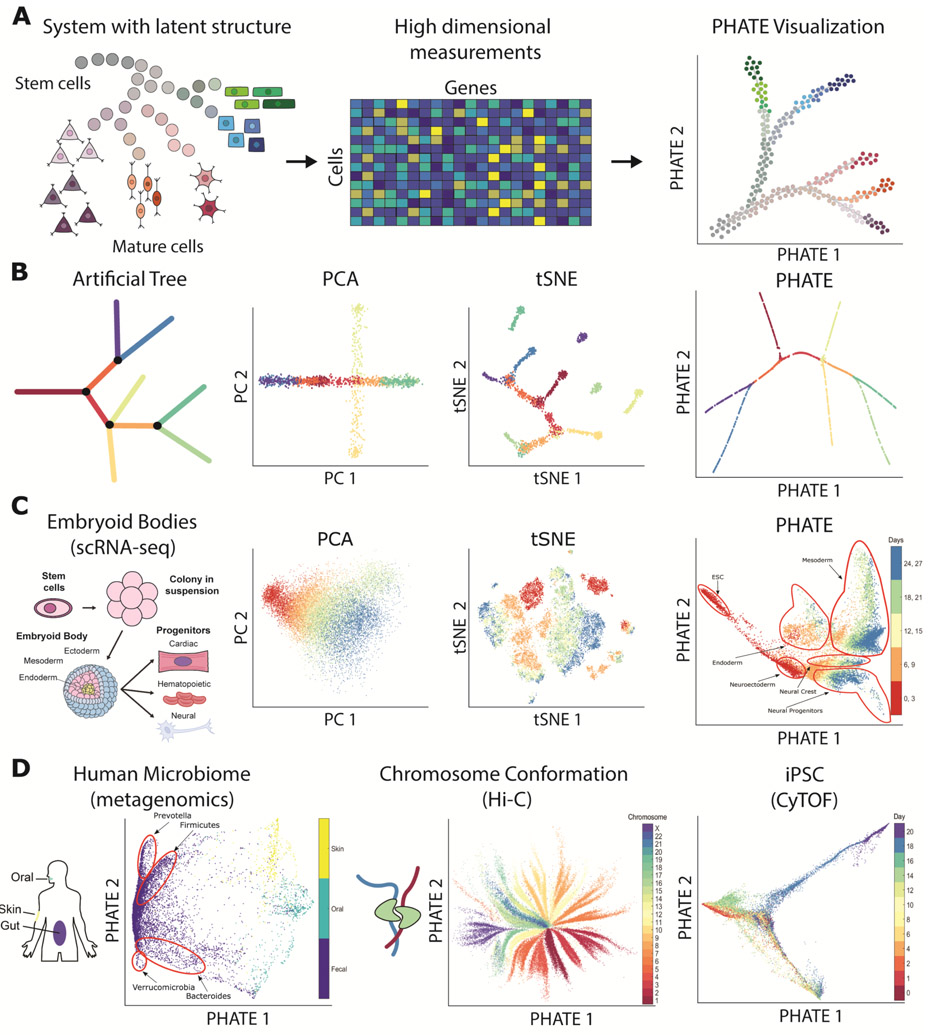
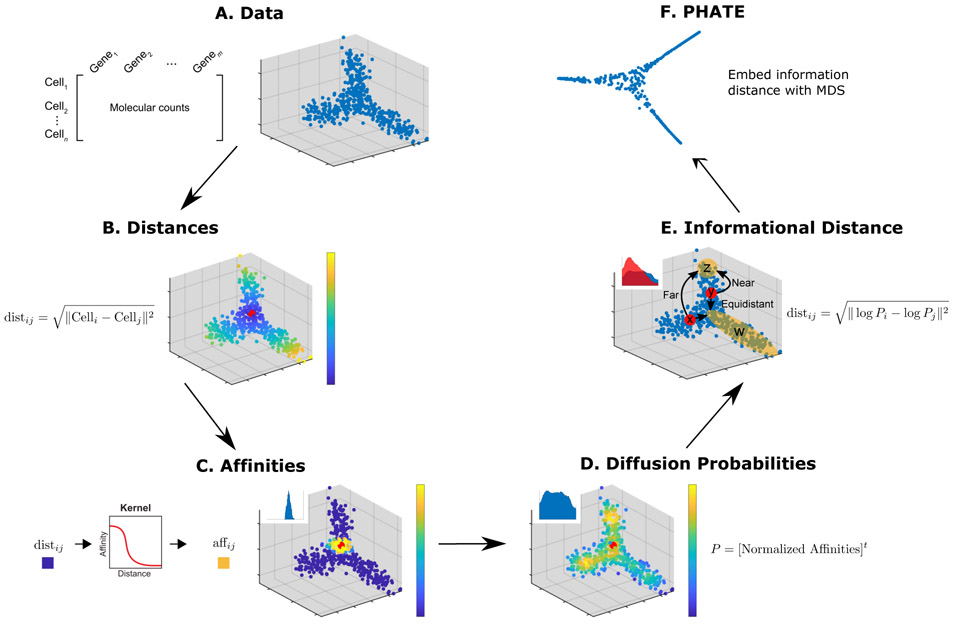
The high-dimensional data created by high-throughput technologies require visualization tools that reveal data structure and patterns in an intuitive form. We present PHATE, a visualization method that captures both local and global nonlinear structure using an information-geometric distance between data points. We compare PHATE to other tools on a variety of artificial and biological datasets, and find that it consistently preserves a range of patterns in data, including continual progressions, branches and clusters, better than other tools. We define a manifold preservation metric, which we call denoised embedding manifold preservation (DEMaP), and show that PHATE produces lower-dimensional embeddings that are quantitatively better denoised as compared to existing visualization methods. An analysis of a newly generated single-cell RNA sequencing dataset on human germ-layer differentiation demonstrates how PHATE reveals unique biological insight into the main developmental branches, including identification of three previously undescribed subpopulations. We also show that PHATE is applicable to a wide variety of data types, including mass cytometry, single-cell RNA sequencing, Hi-C and gut microbiome data.
Figures




References
-
- Maaten L. v. d. and Hinton G, “Visualizing data using t-SNE,” Journal of Machine Learning Research, vol. 9, pp. 2579–2605, 2008.
-
- Amir E.-a. D., Davis KL, Tadmor MD, Simonds EF, Levine JH, Bendall SC, Shenfeld DK, Krishnaswamy S, Nolan GP, and Pe’er D, “viSNE enables visualization of high dimensional single-cell data and reveals phenotypic heterogeneity of leukemia,” Nature Biotechnology, vol. 31, no. 6, pp. 545–552, 2013. - PMC - PubMed
-
- Tenenbaum JB, De Silva V, and Langford JC, “A global geometric framework for nonlinear dimensionality reduction,” Science, vol. 290, no. 5500, pp. 2319–2323, 2000. - PubMed
-
- Becht E, McInnes L, Healy J, Dutertre C-A, Kwok IW, Ng LG, Ginhoux F, and Newell EW, “Dimensionality reduction for visualizing single-cell data using UMAP,” Nature Biotechnology, vol. 37, no. 1, p. 38, 2019. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
