The Escherichia coli transcriptome mostly consists of independently regulated modules
- PMID: 31797920
- PMCID: PMC6892915
- DOI: 10.1038/s41467-019-13483-w
The Escherichia coli transcriptome mostly consists of independently regulated modules
Abstract
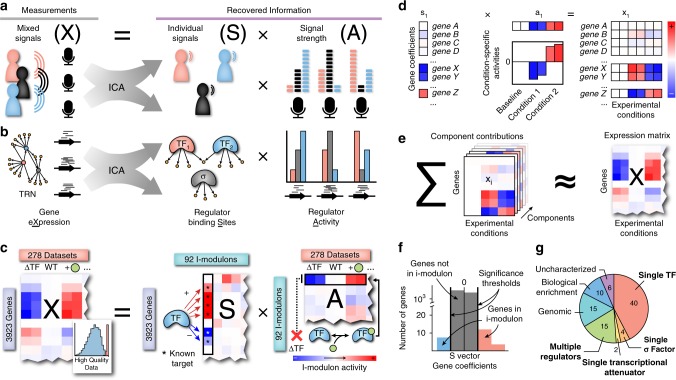
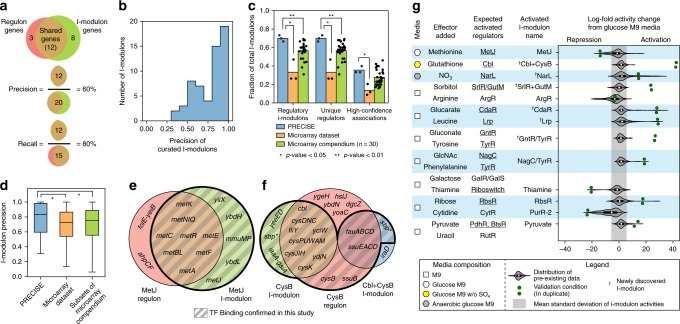
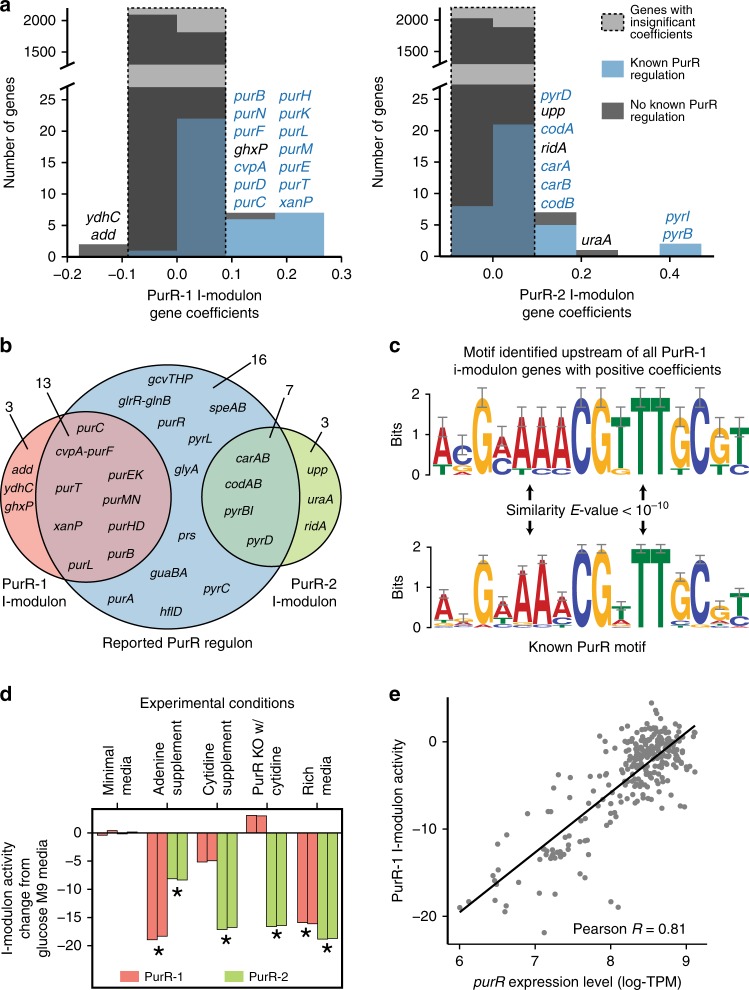
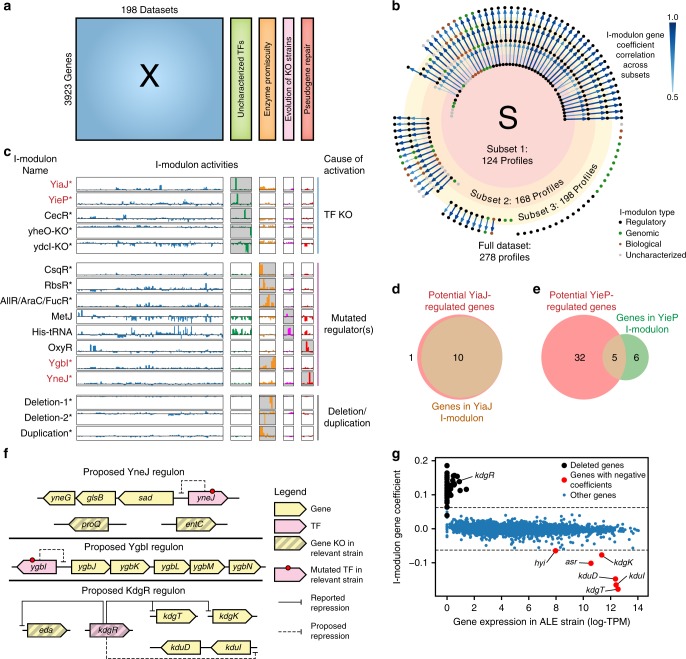
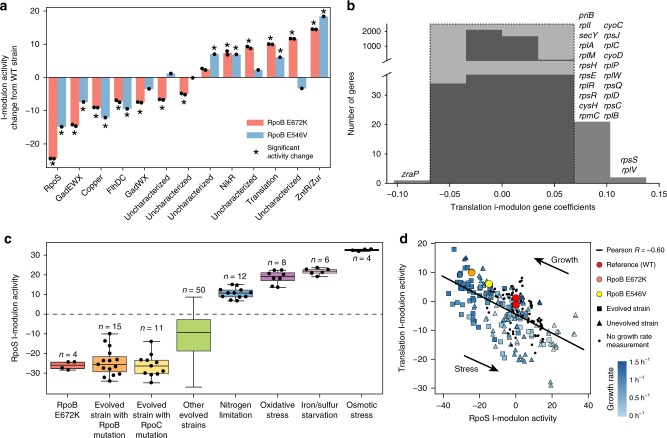
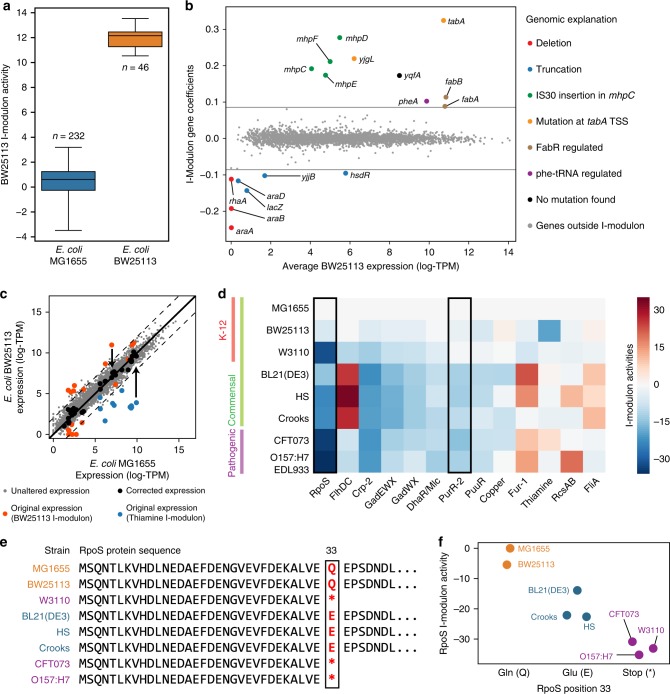
Underlying cellular responses is a transcriptional regulatory network (TRN) that modulates gene expression. A useful description of the TRN would decompose the transcriptome into targeted effects of individual transcriptional regulators. Here, we apply unsupervised machine learning to a diverse compendium of over 250 high-quality Escherichia coli RNA-seq datasets to identify 92 statistically independent signals that modulate the expression of specific gene sets. We show that 61 of these transcriptomic signals represent the effects of currently characterized transcriptional regulators. Condition-specific activation of signals is validated by exposure of E. coli to new environmental conditions. The resulting decomposition of the transcriptome provides: a mechanistic, systems-level, network-based explanation of responses to environmental and genetic perturbations; a guide to gene and regulator function discovery; and a basis for characterizing transcriptomic differences in multiple strains. Taken together, our results show that signal summation describes the composition of a model prokaryotic transcriptome.
Conflict of interest statement
The authors declare no competing interests.
Figures