

Internalized GPCRs as Potential Therapeutic Targets for the Management of Pain
- PMID: 31798411
- PMCID: PMC6874167
- DOI: 10.3389/fnmol.2019.00273
Internalized GPCRs as Potential Therapeutic Targets for the Management of Pain
Abstract
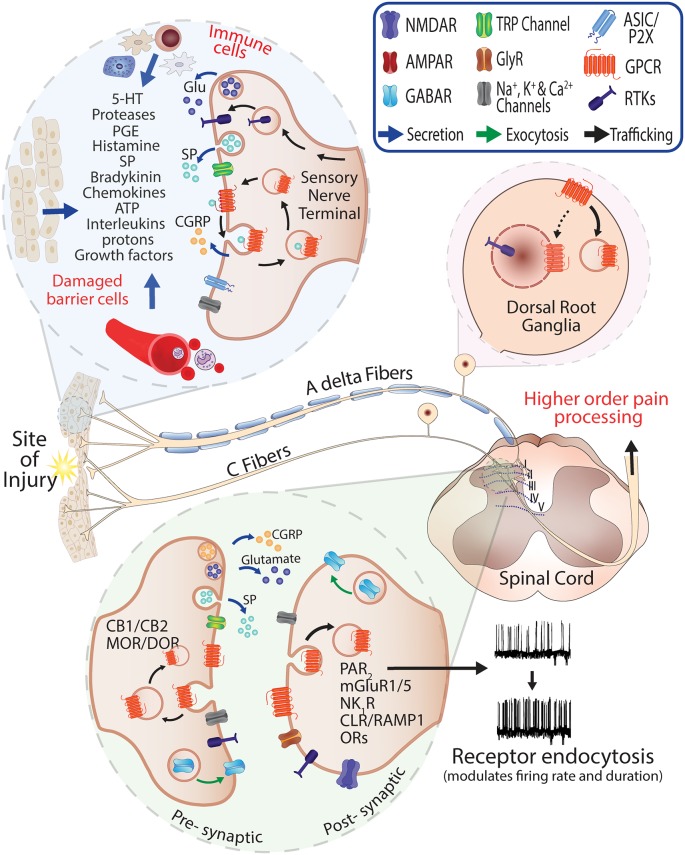
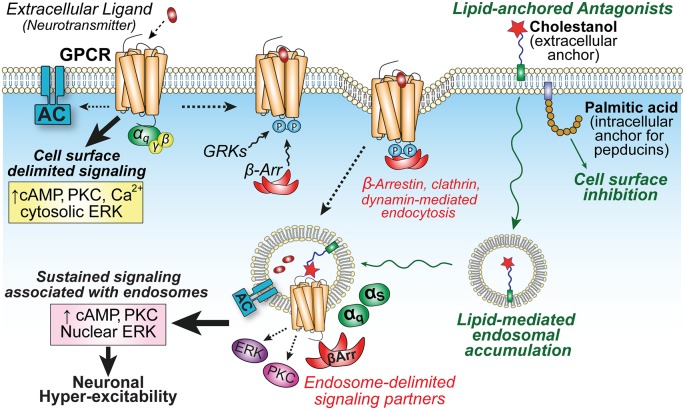
Peripheral and central neurons in the pain pathway are well equipped to detect and respond to extracellular stimuli such as pro-inflammatory mediators and neurotransmitters through the cell surface expression of receptors that can mediate rapid intracellular signaling. Following injury or infection, activation of cell surface G protein-coupled receptors (GPCRs) initiates cell signaling processes that lead to the generation of action potentials in neurons or inflammatory responses such as cytokine secretion by immune cells. However, it is now appreciated that cell surface events alone may not be sufficient for all receptors to generate their complete signaling repertoire. Following an initial wave of signaling at the cell surface, active GPCRs can engage with endocytic proteins such as the adaptor protein β-arrestin (βArr) to promote clathrin-mediated internalization. Classically, βArr-mediated internalization of GPCRs was hypothesized to terminate signaling, yet for multiple GPCRs known to contribute to pain, it has been demonstrated that endocytosis can also promote a unique "second wave" of signaling from intracellular membranes, including those of endosomes and the Golgi, that is spatiotemporally distinct from initial cell-surface events. In the context of pain, understanding the cellular and molecular mechanisms that drive spatiotemporal signaling of GPCRs is invaluable for understanding how pain occurs and persists, and how current analgesics achieve efficacy or promote side-effects. This review article discusses the importance of receptor localization for signaling outcomes of pro- and anti-nociceptive GPCRs, and new analgesic opportunities emerging through the development of "location-biased" ligands that favor binding with intracellular GPCR populations.
Keywords: GPCR; analgesia; drug delivery; endosome; pain; signal transduction; trafficking.
Copyright © 2019 Retamal, Ramírez-García, Shenoy, Poole and Veldhuis.
Figures
References
-
- Abbadie C., Trafton J., Liu H., Mantyh P. W., Basbaum A. I. (1997). Inflammation increases the distribution of dorsal horn neurons that internalize the neurokinin-1 receptor in response to noxious and non-noxious stimulation. J. Neurosci. 17, 8049–8060. 10.1523/JNEUROSCI.17-20-08049.1997 - DOI - PMC - PubMed
LinkOut - more resources
Full Text Sources